Publications
Self-similar solution for laminar bubbly flow evolving from a vertical plate
The development of a bubble plume from a vertical gas-evolving electrode is driven by buoyancy and hydrodynamic bubble dispersion. This canonical fluid mechanics problem is relevant for both thermal and electrochemical processes. We adopt a mixture model formulation for the two-phase flow, considering variable density (beyond Boussinesq), viscosity and hydrodynamic bubble dispersion. Introducing a new change of coordinates, inspired by the Lees–Dorodnitsyn transformation, we obtain a new self-similar solution for the laminar boundary layer equations. The results predict a wall gas fraction and gas plume thickness that increase with height to the power of 1/5 before asymptotically reaching unity and scaling with height to the power 2/5, respectively. The vertical velocity scales with height to the power of 3/5. Our analysis shows that self-similarity is only possible if gas conservation is entirely formulated in terms of the gas specific volume instead of the gas fraction.
N. Valle and J.W. Haverkort
Nanofluidic ion-exchange membranes: Can their conductance compete with polymeric ion-exchange membranes?
Nanofluidic membranes (NFMs) are gaining prominence as alternative ion-exchange membranes, because of their distinct selectivity mechanism, which does not rely on functional groups on a polymeric backbone but rather on charged nanopores that allow straight ion-conductive pathways for efficient ion transport. We measured the conductivity of commercial anodized aluminum oxide membranes with different pore sizes under different current densities and electrolyte concentrations. We also simulated a nanopore channel with charged walls between two electrolyte reservoirs. Our findings indicate that electrolyte concentration is the main parameter that determines NFM conductivity, with a linear dependence at least up to 1 M. Our study shows that the optimal pore length is between 0.5 and 5 μm considering the trade-off between selectivity and conductance. On the other hand, the conductance is not sensitive to the pore diameter. Conical nanopores are a way to increase conductance, but according to our results, this increase comes at the expense of selectivity. Our findings suggest that NFMs can outperform polymeric ion-exchange membranes in certain electrochemical applications, such as reverse electrodialysis, but not in applications that use low electrolyte concentrations on both sides of the membrane.
Petrov, K. V., Hurkmans, J. W., Hartkamp, R., & Vermaas, D. A.

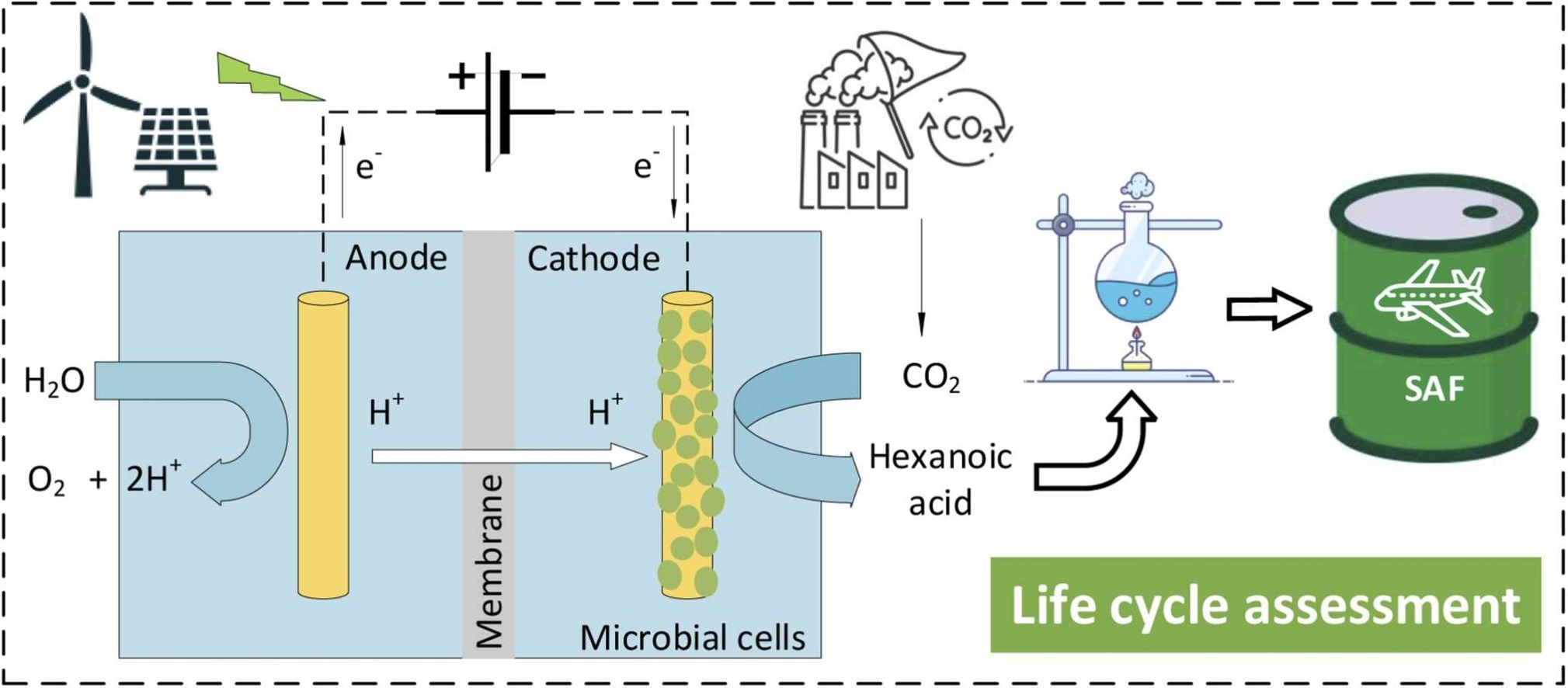
Life cycle assessment of hexanoic acid production via microbial electrosynthesis and renewable electricity: Future opportunities
Microbial electrosynthesis (MES) is a novel carbon utilisation technology aiming to contribute to a circular economy by converting CO2 and renewable electricity into value-added chemicals. This study presents a cradle-to-gate life cycle assessment (LCA) of hexanoic acid (C6A) production using MES, comparing this production with alternative technologies. It also includes a cradle-to-grave LCA for potentially converting C6A into a neat sustainable aviation fuel (SAF). On a cradle-to-gate basis, MES-based C6A exhibits a carbon footprint at 5.5 t CO2eq/tC6A, similar to fermentation- and plant-based C6A. However, its direct land use is more than one order of magnitude lower than plant-based C6A. On a cradle-to-grave basis, MES-based neat SAF emits 325 g CO2eq/MJ neat SAF, which is significantly higher than the counterparts from currently certified routes and conventional petroleum-derived jet fuel. However, its negligible indirect land use change emissions might potentially make it competitive against neat SAFs originating from first-generation biomass.
Jisiwei Luo, Mar Pérez-Fortes, Adrie J.J. Straathof, Andrea Ramirez
Sustainable design of multiscale CO2 electrolysis: A value sensitive design-based approach
The present study utilizes a value sensitive design (VSD) inspired approach to contribute to the design and implementation of CO2 electrolysis (CO2E) within the framework of carbon capture and utilization (CCU) technologies, which convert CO2 into valuable products. The focus of this study is on a low technology readiness level (TRL) technology, yet likely relevant to reach climate neutrality by 2050. We examine the perspectives of stakeholders along the supply chain and proactively identify relevant sustainability-related values and potential conflicts among them. Thus the current work highlights the importance of considering a broad range of stakeholders and their values in the early stages of technological design. The research approach is consisting of various steps inspired by value sensitive design (VSD): identifying relevant values and norms associated with CO2 electrolysis through literature analysis, conducting qualitative interviews with relevant stakeholders to triangulate the results. Subsequently, a value-based alignment network analysis was employed to examine shared values that are central for the design of the technology. The findings indicate that sustainability-related values such as concern for nature, climate change mitigation, the use of renewable energy, critical raw materials, cost, and return on investment, albeit with potential differences in interpretation, are increasingly becoming central considerations in the decision-making processes of individuals, businesses, and governments alike. Based on these findings, specific aspects of technology design, namely scale, location, integration, and synthesized product, that can impact a wide range of identified values, are discussed.
Marula Tsagkari, Ibo van de Poel, Mar Pérez-Fortes
Earlier publications
-
Eliminating redox-mediated electron transfer mechanisms on a supported molecular catalyst enables CO2 conversion to ethanol
Molecular catalysts play a significant role in chemical transformations, utilizing changes in redox states to facilitate reactions. To date molecular electrocatalysts have efficiently produced single-carbon products from CO2 but have struggled to achieve a carbon–carbon coupling step. Conversely, copper catalysts can enable carbon–carbon coupling, but lead to broad C2+ product spectra. Here we subvert the traditional redox-mediated reaction mechanisms of organometallic compounds through a heterogeneous nickel-supported iron tetraphenylporphyrin electrocatalyst, facilitating electrochemical carbon–carbon coupling to produce ethanol. This represents a marked behavioural shift compared with carbon-supported metalloporphyrins. Extending the approach to a three-dimensional porous nickel support with adsorbed iron tetraphenylporphyrin, we attain ethanol Faradaic efficiencies of 68% ± 3.2% at −0.3 V versus a reversible hydrogen electrode (pH 7.7) with partial ethanol current densities of −21 mA cm−2. Separately we demonstrate maintained ethanol production over 60 h of operation. Further consideration of the wide parameter space of molecular catalyst and metal electrodes shows promise for additional chemistries and achievable metrics.
M Abdinejad, A Farzi, R Möller-Gulland, F Mulder, C Liu, J Shao, M Robert, A Seifitokaldani, T Burdyny
-
Carbon-based products are essential to society, yet producing them from fossil fuels is unsustainable. Microorganisms have the ability to take up electrons from solid electrodes and convert carbon dioxide (CO2) to valuable carbon-based chemicals. However, higher productivities and energy efficiencies are needed to reach a viability that can make the technology transformative. Here, we show how a biofilm-based microbial porous cathode in a directed flow-through electrochemical system can continuously reduce CO2 to even-chain C2–C6 carboxylic acids over 248 days. We demonstrate a threefold higher biofilm concentration, volumetric current density, and productivity compared with the state of the art. Most notably, the volumetric productivity (VP) resembles those achieved in laboratory-scale and industrial syngas (CO-H2-CO2) fermentation and chain elongation fermentation. This work highlights key design parameters for efficient electricity-driven microbial CO2 reduction. There is need and room to improve the rates of electrode colonization and microbe-specific kinetics to scale up the technology.
Oriol Cabau-Peinado, Marijn Winkelhorst, Rozanne Stroek, Kunal Masania, Jean Marc Daran, Ludovic Jourdin
-
Electrochemical CO2 reduction aims to compete with Power-to-X alternatives but is well behind the scales of water electrolyzers and thermochemical reactors. In a recent issue of Nature Chemical Engineering, Crandall and co-workers demonstrate a 1000 cm2 tandem CO2/CO electrolyzer for acetate production. The work invites discussion on scientific and engineering scale-up challenges.
Thomas Burdyny, Fokko M. Mulder
-
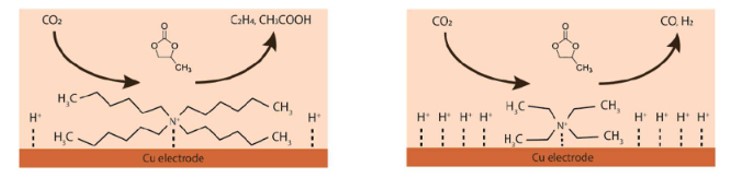
Electrochemical CO2 reduction in non-aqueous solvents is promising due to the increased CO2 solubility of organic-based electrolytes compared to aqueous electrolytes. Here the effect of nine different salts in propylene carbonate (PC) on the CO2 reduction product distribution of polycrystalline Cu is investigated. Three different cations (tetraethylammonium (TEA), tetrabutylammonium (TBA), and tetrahexylammonium (THA)) and three different anions (chloride (Cl), tetrafluoroborate (BF4), and hexafluorophosphate (PF6)) were used. Chronoamperometry and in-situ FTIR measurements show that the size of the cation has a crucial role in the selectivity. A more hydrophobic surface is obtained when employing a larger cation with a weaker hydration shell. This stabilizes the CO2-· radical and promotes the formation of ethylene. CO2 reduction in 0.7 M THACl/PC shows the highest hydrocarbon formation. Lastly, we hypothesize that the hydrocarbon formation pathway is not through C-C coupling, as the CO solubility in PC is very high, but through the dimerization of the COH intermediate.
Iris Burgers, Boris Wortmann, Amanda C. Garcia, Connor Deacon-Price, Elena Pérez-Gallent, Earl Goetheer, Ruud Kortlever
-
In this study, the effect of halide anions on the selectivity of the CO2 reduction reaction to CO was investigated in choline-based ethylene glycol solutions containing different halides (ChCl : EG, ChBr : EG, ChI : EG). The CO2RR was studied using silver (Ag) and gold (Au) electrodes in a compact H-cell. Our findings reveal that chloride effectively suppresses the hydrogen evolution reaction and enhances the selectivity of carbon monoxide production on both Ag and Au electrodes, with relatively high selectivity values of 84 % and 62 %, respectively. Additionally, the effect of varying ethylene glycol content in the choline chloride-containing electrolyte (ChCl : EG 1 : X, X=2, 3, 4) was investigated to improve the current density during CO2RR on the Ag electrode. We observed that a mole ratio of 1 : 4 exhibited the highest current density with a comparable faradaic efficiency toward CO. Notably, an evident surface reconstruction process took place on the Ag surface in the presence of Cl− ions, whereas on Au, this phenomenon was less pronounced. Overall, this study provides new insights into anion-induced surface restructuring of Ag and Au electrodes during CO2RR, and its consequences on the reduction performance on such surfaces in non-aqueous electrolytes.
Hengameh Farahmandazad, Simone Asperti, Ruud Kortlever, Earl Goetheer, Wiebren de Jong
-
To successfully transition from a fossil fuel-based society to one based on renewable energy, switching only to renewable energy sources is not enough. New, renewable processes and catalyst materials to synthesize bulk chemicals and transportation fuels will need to be developed. Electrochemical CO2 reduction is a promising renewable process as it directly uses renewable energy to produce the desired chemicals under mild conditions.
Despite many efforts to find better catalytic materials, progress remains slow due to product quantification methods with high detection limits. In an effort to accelerate catalyst development research, we have designed an improved DEMS setup that is four times faster than conventional research in H cells. To the best of our knowledge, it is the first DEMS setup that is able to simultaneously quantify carbon monoxide and liquid products without the need for additional measurements.
Daniël van den Berg, Hendrik Paul Lopuhaä, Ruud Kortlever
-
Carbon dioxide (CO2) electrolysis on copper (Cu) catalysts has attracted interest due to its direct production of C2+ feedstocks. Using the knowledge that CO2 reduction on copper is primarily a tandem reaction of CO2 to CO and CO to C2+ products, we show that modulating CO concentrations within the liquid catalyst layer allows for a C2+ selectivity of >80% at 200 mA cm−2 under broad conversion conditions. The importance of CO pooling is demonstrated through residence time distribution curves, varying flow fields (serpentine/parallel/interdigitated), and flow rates. While serpentine flow fields require high conversions to limit CO selectivity and maximize C2+ selectivity, the longer CO residence times of parallel flow fields achieve similar selectivity over broad flow rates. Critically, we show that parts of the catalyst area predominantly reduce CO instead of CO2 as supported by CO reduction experiments, transport modelling, and achieving a CO2 utilization efficiency greater than the theoretical limit of 25% for C2+ products.
S Subramanian, J Kok, P Gholkar, A Sajeev Kumar, HP Iglesias van Montfort, R Kortlever, A Urakawa, B Dam, T Burdyny
-
Electrochemical oxygen reduction is a promising and sustainable alternative to the current industrial production method for hydrogen peroxide (H2O2), which is a green oxidant in many (emerging) applications in the chemical industry, water treatment, and fuel cells. Low solubility of O2 in water causes severe mass transfer limitations and loss of H2O2 selectivity at industrially relevant current densities, complicating the development of practical-scale electrochemical H2O2 synthesis systems. We tested a flow-by and flow-through configuration and suspension electrodes in an electrochemical flow cell to investigate the influence of electrode configuration and flow conditions on mass transfer and H2O2 production. We monitored the H2O2 production using Cu-tmpa (tmpa = tris(2-pyridylmethyl)amine) as a homogeneous copper-based catalyst in a pH-neutral phosphate buffer during 1 h of catalysis and estimated the limiting current density from CV scans. We achieve the highest H2O2 production and a 15–20 times higher geometrical limiting current density in the flow-through configuration compared to the flow-by configuration due to the increased surface area and foam structure that improved mass transfer. The activated carbon (AC) material in suspension electrodes, which have an even larger surface area, decomposes all produced H2O2 and proves unsuitable for H2O2 synthesis. Although the mass transfer limitations seem to be alleviated on the microscale in the flow-through system, the high O2 consumption and H2O2 production cause challenges in maintaining the initially reached current density and Faradaic efficiency (FE). The decreasing ratio between the concentrations of the O2 and H2O2 in the bulk electrolyte will likely pose a challenge when proceeding to larger systems with longer electrodes. Tuning the reactor design and operating conditions will be essential in maximizing the FE and current density.
Ligthart, N. E., van Langevelde, P. H., Padding, J. T., Hetterscheid, D. G., & Vermaas, D. A.
-
CO2 electrolysis allows the sustainable production of carbon-based fuels and chemicals. However, state-of-the-art CO2 electrolysers employing anion exchange membranes (AEMs) suffer from (bi)carbonate crossover, causing low CO2 utilization and limiting anode choices to those based on precious metals. Here we argue that bipolar membranes (BPMs) could become the primary option for intrinsically stable and efficient CO2 electrolysis without the use of scarce metals. Although both reverse- and forward-bias BPMs can inhibit CO2 crossover, forward-bias BPMs fail to solve the rare-earth metals requirement at the anode. Unfortunately, reverse-bias BPM systems presently exhibit comparatively lower Faradaic efficiencies and higher cell voltages than AEM-based systems. We argue that these performance challenges can be overcome by focusing research on optimizing the catalyst, reaction microenvironment and alkali cation availability. Furthermore, BPMs can be improved by using thinner layers and a suitable water dissociation catalyst, thus alleviating core remaining challenges in CO2 electrolysis to bring this technology to the industrial scale.
K Petrov, C Koopman, S Subramanian, M Koper, T Burdyny, D Vermaas
-
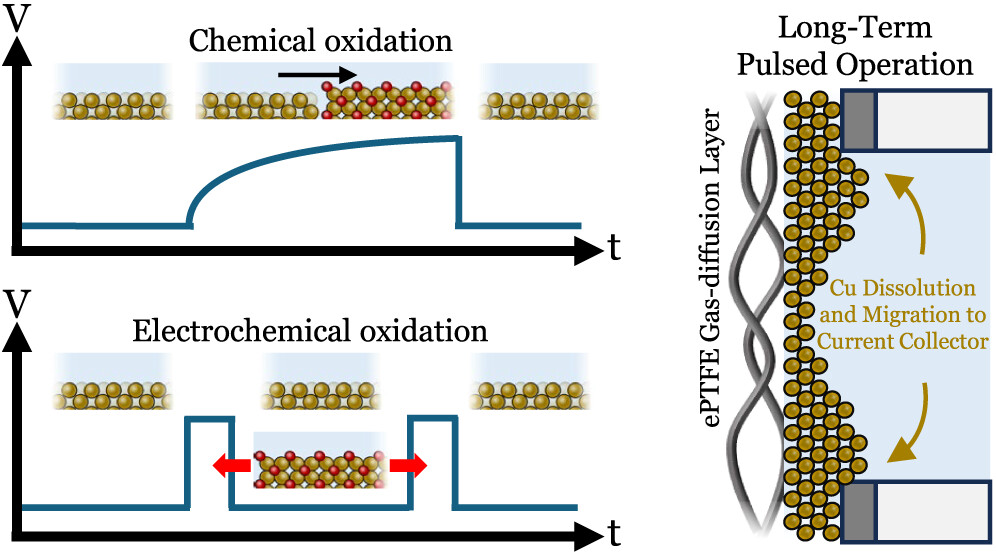
Using copper (Cu) as an electrocatalyst uniquely produces multicarbon products (C2+-products) during the CO2 reduction reaction (CO2RR). However, the CO2RR stability of Cu is presently 3 orders of magnitude shorter than required for commercial operation. One means of substantially increasing Cu catalyst lifetimes is through periodic oxidative processes, such as cathodic–anodic pulsing. Despite 100-fold improvements, these oxidative methods only delay, but do not circumvent, degradation. Here, we provide an interrogation of chemical and electrochemical Cu oxidative processes to identify the mechanistic processes leading to stable CO2RR through electrochemical and in situ Raman spectroscopy measurements. We first examine chemical oxidation using an open-circuit potential (OCP), identifying that copper oxidation is regulated by the transient behavior of the OCP curve and limited by the rate of the oxygen reduction reaction (ORR). Increasing O2 flux to the cathode subsequently increased ORR rates, both extending lifetimes and reducing “off” times by 3-fold. In a separate approach, the formation of Cu2O is achieved through electrochemical oxidation. Here, we establish the minimum electrode potentials required for fast Cu oxidation (−0.28 V vs Ag/AgCl, 1 M KHCO3) by accounting for transient local pH changes and tracking oxidation charge transfer. Lastly, we performed a stability test resulting in a 20-fold increase in stable ethylene production versus the continuous case, finding that spatial copper migration is slowed but not mitigated by oxidative pulsing approaches alone.
J Kok, J de Ruiter, W van der Stam, T Burdyny





-
Combining intermittent renewable electricity (IRE) with carbon capture and utilisation is urgently needed in the chemical sector. In this context, microbial electrosynthesis (MES) has gained attention. It can electrochemically produce hexanoic acid, a value-added chemical, from CO2. However, there is a lack of understanding regarding how the intermittency of renewable electricity could impact the design of a MES plant. We studied this using Aspen Plus models.
A MES plant that was powered by constant grid electricity could operate from 100% down to 70% of its nominal capacity, at which point the heat exchangers and the internal geometrical design of the distillation towers became bottlenecks. The levelised production cost of hexanoic acid (LPCC6A) was estimated at 4.0 €/kg. Switching to IRE supply increased LPCC6A to 5.3 €/kg (for wind electricity) and 4.7 €/kg (for hybrid renewable electricity).
A battery energy storage system (BESS) was deployed. The lowest LPCC6A was found at a BESS installation of 29 GJ/h for wind electricity (5.1 €/kg) and at 12 GJ/h for hybrid renewable electricity (4.7 €/kg). In both situations, the volume flexibility of the MES plant was not improved. At the investigated market and operating conditions, coupling IRE to the MES plant was economically infeasible.
Jisiwei Luo, Mar Pérez-Fortes, Paola Ibarra-Gonzalez, Adrie J.J. Straathof, Andrea Ramirez
-
Covalent organic frameworks (COFs) are ideal platforms to spatially control the integration of multiple molecular motifs throughout a single nanoporous framework. Despite this design flexibility, COFs are typically synthesized using only two monomers. One bears the functional motif for the envisioned application, while the other is used as an inert connecting building block. Integrating more than one functional motif extends the functionality of COFs immensely, which is particularly useful for multistep reactions such as electrochemical reduction of CO2. In this systematic study, we synthesized five Ni(II)- and Zn(II)-porphyrin-based COFs, including two pure component COFs (Ni100 and Zn100) and three mixed Ni/Zn-COFs (Ni75/Zn25, Ni50/Zn50, and Ni25/Zn75). Among these, the Ni50/Zn50-COF exhibited the highest catalytic performance for the electroreduction of CO2 to CO and formate at −0.6 V vs RHE, as was observed in an H-cell. The catalytic performance of the COF catalysts was further extended to a zero-gap membrane electrode assembly (MEA) operation where, utilizing Ni50/Zn50, CH4 was detected along with CO and formate at a high current density of 150 mA cm–2. In contrast, under these conditions predominantly H2 and CO were detected at Ni100 and Zn100 respectively, indicating a clear synergistic effect between the Ni- and Zn-porphyrin units.
Hugo Veldhuizen, Maryam Abdinejad, Pieter J. Gilissen, Jelco Albertsma, Thomas Burdyny, Frans D. Tichelaar, Sybrand van der Zwaag, and Monique A. van der Veen

-
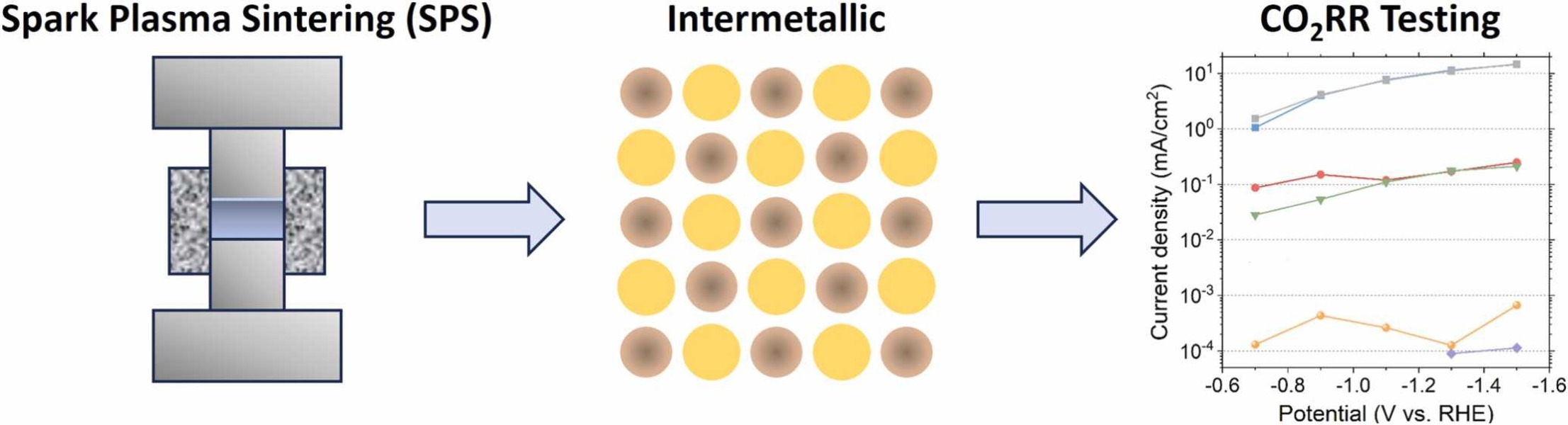
In this study, we experimentally screen a promising class of intermetallic alloys for the electrochemical reduction of CO2 toward hydrocarbon products. Based on previous DFT-based screening papers, combinations of strongly CO-binding metals such as iron, cobalt, and nickel with weakly CO-binding metals such as gallium, aluminium or zinc were selected as potentially promising catalytic materials. Despite the challenging production of these alloys, we report a general two-step synthesis method for intermetallic alloys and discuss the specific synthesis conditions that must be taken into account when synthesising these materials. After their synthesis, we use a recently developed differential electrochemical mass spectrometry (DEMS) setup to rapidly quantify the CO2 reduction products over a range of potentials. Almost all newly developed intermetallic catalysts are shown to produce methane and ethylene, while the CoSn catalyst showed higher selectivity towards formate production. However, all tested catalysts mostly produced hydrogen and only reduce CO2 to a small extent, despite the favourable computational screening results. We discuss possible reasons for this discrepancy and outline a more holistic approach for linking future DFT studies with experiments.
Daniël van den Berg, Johannes C. Brouwer, Ruud W.A. Hendrikx, Ruud Kortlever
-
The anodic co-production of hydrogen peroxide (H2O2) during alkaline water electrolysis has gained interest as a sustainable alternative for anthraquinone oxidation. However, electrochemical H2O2 production is often studied with idealized laboratory setups to determine the H2O2 formation kinetics. In this work, we perform the reaction with industrially relevant operating principles using a flow cell with separately recirculating anolyte and catholyte. We then fit the data to an analytical model that we derive based on mole balances that accounts for anodic generation, anodic oxidation, and bulk disproportionation of H2O2, as well as electrolyte volumes and electrode surface area. We performed experiments at 100, 200, and 300 mA cm-2 to derive values for the reaction system. At 200 mA cm-2, we found a generation rate of 0.037 mmol min-1 cm-2 (FEH2O2 = 59%) and an anodic decomposition rate constant of 0.304 cm min-1, with a bulk disproportionation rate constant of 1.85 x 10-3 min-1. We successfully applied our model to two sources in literature to derive values for their systems as well. In all cases, the contribution of anodic oxidation of H2O2 was found to be the larger loss mechanism in comparison to bulk disproportionation. Using the analytical model, we show that decreasing the reservoir volume is a simple way to increase the H2O2 concentration over time. Further refinement of the model can be achieved through the use of mass transfer relationships based on electrolyzer geometries to describe the anodic oxidation of H2O2 in the mole balance equations.
Sohan A. Phadke, Wiebren de Jong, J.W. Haverkort

-
Poor mass transport to or from vertical gas-evolving electrodes can adversely impact energy efficiency and product purity in the production of hydrogen, chlorine, and various metals. A proper description that combines natural convection with micromixing of growing, coalescing, and departing bubbles is presently lacking. This work develops a simple, physically sound analytical model that includes the influence of bubble size, flow regime, and bubble surface coverage. By comprehensively reviewing mass transfer measurements from the water electrolysis literature, we observe that the surface coverage of oxygen bubbles increases much more strongly with increasing current density than an often-used square root scaling predicts. Strong differences are observed in the degree of micromixing of hydrogen and oxygen bubbles in alkaline and acidic electrolytes. These varied results can all be explained by a combination of electrocapillarity, and coalescence induced by either a high surface coverage or Marangoni flows.
J.W. Haverkort

-
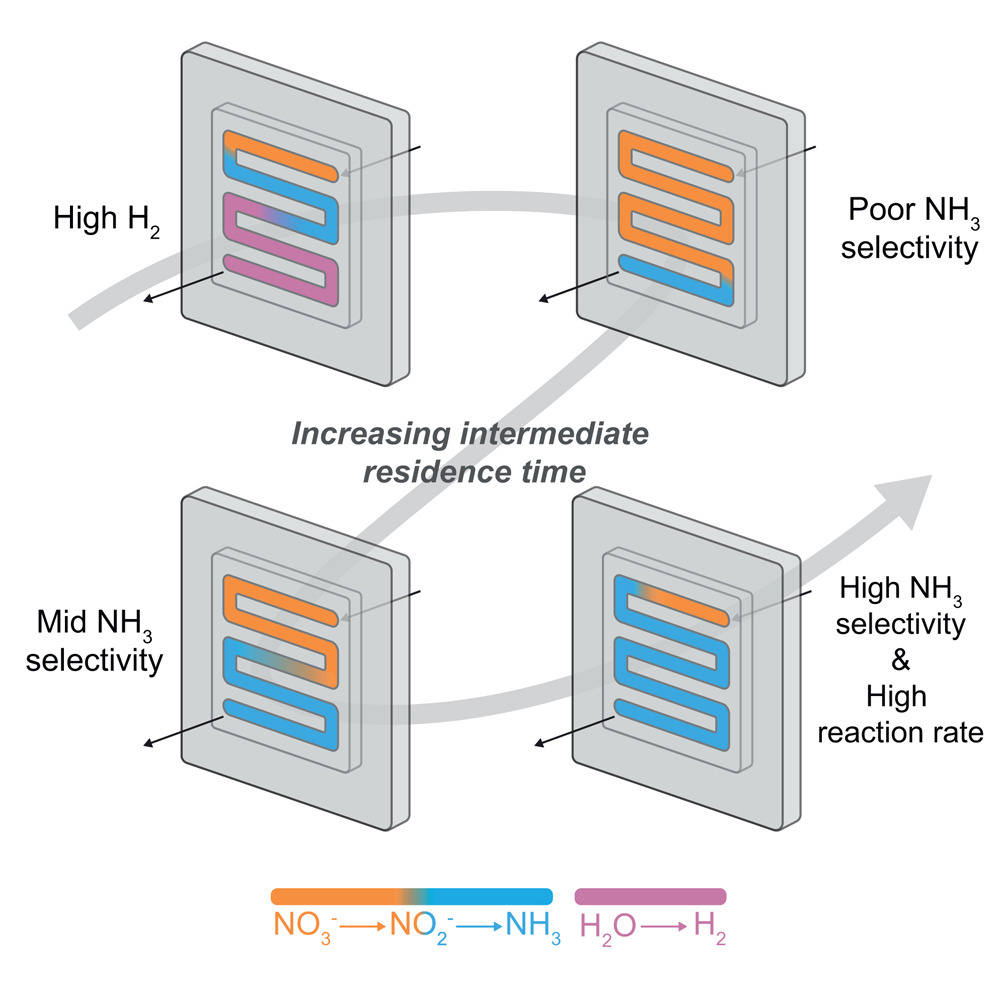
Electrochemical ammonia (NH3) synthesis from nitrate (NO3−) offers a promising greener alternative to the fossil-fuel-based Haber-Bosch process to support the increasing demand for nitrogen fertilizers while removing environmental waste. Previous studies have mainly focused on designing catalysts to promote the direct conversion (NO3− → NH3) while suppressing the two-step pathway (NO3− → NO2− → NH3). We hypothesize that efficient nitrate reduction is possible on simple catalysts by instead promoting the two-step reaction and using chemical reactor principles in a membrane electrode assembly, despite NO2− intermediates. Here, we use an unmodified copper catalyst and control reactivity through current density, flow rate, and electrolyte recycling. Balancing the electrolyte flow rate with current density results in ideal residence times for NO2−, allowing for 91% FENH3 in a 5 cm2 electrolyzer with a NO3− to NH3 partial current of 1.8 A. This work shows that traditional engineering principles can substantially boost the NO3 reduction reaction, even for simple catalysts.
T Yuan, M Li, S Subramanian, J Kok, M Li, A Urakawa, O Voznyy, T Burdyny
-
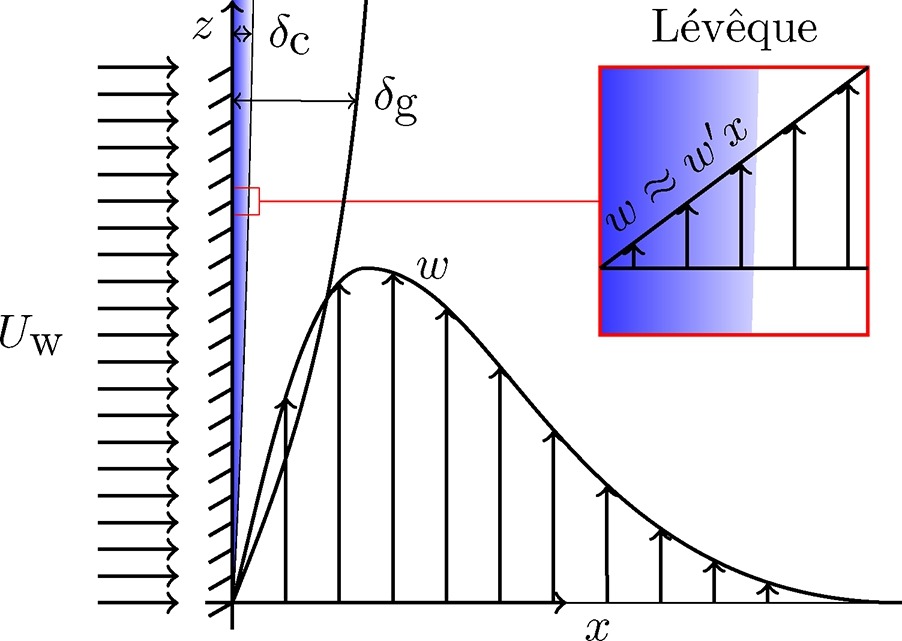
The high mass transfer to or from gas-evolving electrodes is an attractive feature of electrochemical reactors, which can be partly attributed to the large convective flows that arise due to the buoyancy of bubbles. We derive exact analytical expressions for mass transfer coefficients for the case of constant gas flux boundary conditions. For the mass transport both Dirichlet and Neumann boundary conditions are considered. We deploy a recently derived self-similar solution of laminar two-phase flows, with density, hydrodynamic diffusivity, and viscosity dependent on the local gas fraction. Combining this with the Lévêque approximation, new mass transfer coefficients are obtained analytically. These new results are relevant for various electrochemical processes with gas evolution as well as boiling. The new formulation shows the mass transfer coefficient to scale with the vertical coordinate z proportional to z−1/5 for short electrodes and low current densities and z−4/15 for long ones and high current densities. The former limit also applies when buoyancy is due to temperature or concentration differences in the case that density differences are small. We provide a general overview considering all possible gas and mass boundary conditions combinations and a comparison with the Boussinesq approximation of small density differences.
Valle, N., Haverkort, J. W.

-
CO2 from carbonate-based capture solutions requires a substantial energy input. Replacing this step with (bi)carbonate electrolysis has been commonly proposed as an efficient alternative that coproduces CO/syngas. Here, we assess the feasibility of directly integrating air contactors with (bi)carbonate electrolyzers by leveraging process, multiphysics, microkinetic, and technoeconomic models. We show that the copresence of CO32– with HCO3– in the contactor effluent greatly diminishes the electrolyzer performance and eventually results in a reduced CO2 capture fraction to ≤1%. Additionally, we estimate suitable effluents for (bi)carbonate electrolysis to require 5–14 times larger contactors than conventionally needed contactors, leading to unfavorable process economics. Notably, we show that the regeneration of the capture solvent inside (bi)carbonate electrolyzers is insufficient for CO2 recapture. Thus, we suggest process modifications that would allow this route to be operationally feasible. Overall, this work sheds light on the practical operation of integrated direct air capture with (bi)carbonate electrolysis.
R Kas, P Brimley, AM Crow, A Somoza-Tornos, BM Hodge, T Burdyny, and WA Smith

-
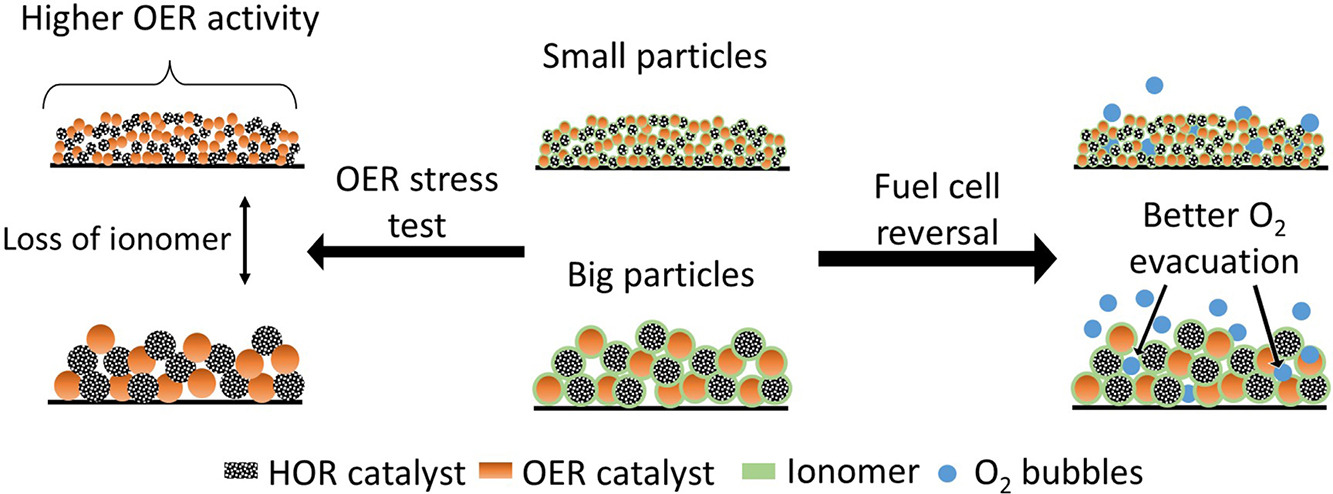
When no hydrogen can reach the Pt catalyst in the anode for the hydrogen oxidation reaction (HOR) of an operating proton exchange membrane fuel cell (PEMFC), the anode potential increases and causes the cell potential to be reversed compared to normal operation conditions. During this reversal, the oxygen evolution reaction (OER) and carbon oxidation reaction (COR) will occur at the anode, where the COR has devastating consequences for the electrode. Introducing an OER catalyst limits the COR to occur, which makes a reversal tolerant anode (RTA). In this research, RTAs are differentiated by applying different ball milling times during catalyst layer processing, forming big and small OER (IrOx/TiOx) and HOR (Pt/C) catalyst particles. The two different particle sizes were electrochemically tested using a rotating disc electrode (RDE). Both catalyst sizes show a decrease in OER activity (mA cm−2) accompanied by loss of the ionomer in a self-developed accelerated stress test (AST). The small particle RTAs show higher OER activity as a result of increased surface area. However, during a chronopotentiometry measurement, which mimics a fuel cell reversal, the small particle coatings show a worse reversal tolerance. This phenomenon can be attributed to the increased difficulty in removing oxygen bubbles.
S.J.T. Homan, K. Aylar, A. Jurjevic, M. Scolari, A. Urakawa, P. Taheri
-
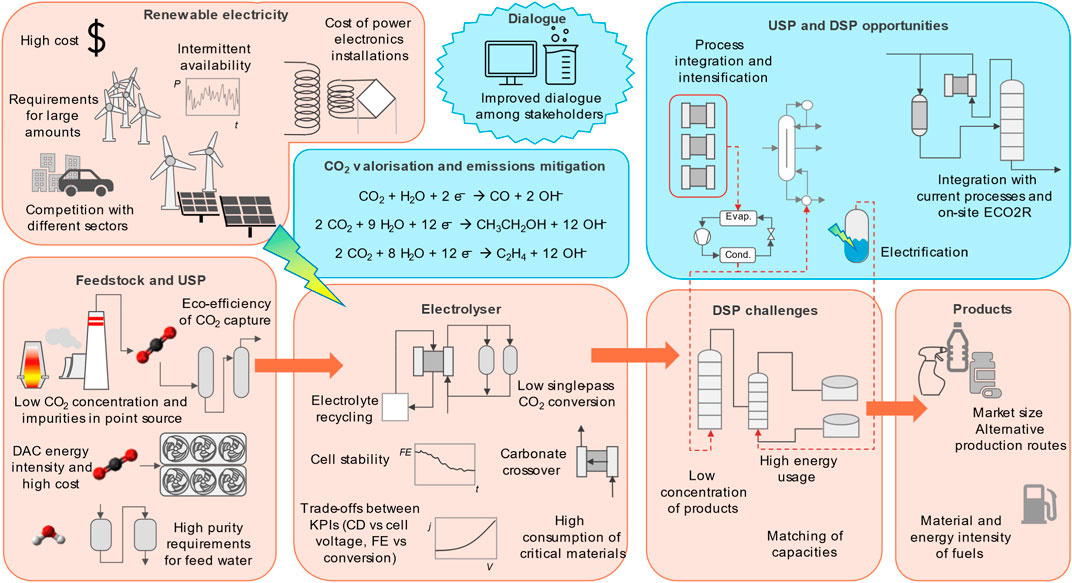
Despite the huge efforts devoted to the development of the electrochemical reduction of CO2 (ECO2R) in the past decade, still many challenges are present, hindering further approaches to industrial applications. This paper gives a perspective on these challenges from a Process Systems Engineering (PSE) standpoint, while at the same time highlighting the opportunities for advancements in the field in the European context. The challenges are connected with: the coupling of these processes with renewable electricity generation; the feedstock (in particular CO2); the processes itself; and the different products that can be obtained. PSE can determine the optimal interactions among the components of such systems, allowing educated decision making in designing the best process configurations under uncertainty and constrains. The opportunities, on the other hand, stem from a stronger collaboration between the PSE and the experimental communities, from the possibility of integrating ECO2R into existing industrial productions and from process-wide optimisation studies, encompassing the whole production cycle of the chemicals to exploit possible synergies.
Riccardo Dal Mas, Ana Somoza-Tornos, Mar Pérez-Fortes, Ruud Kortlever, Anton A. Kiss
-
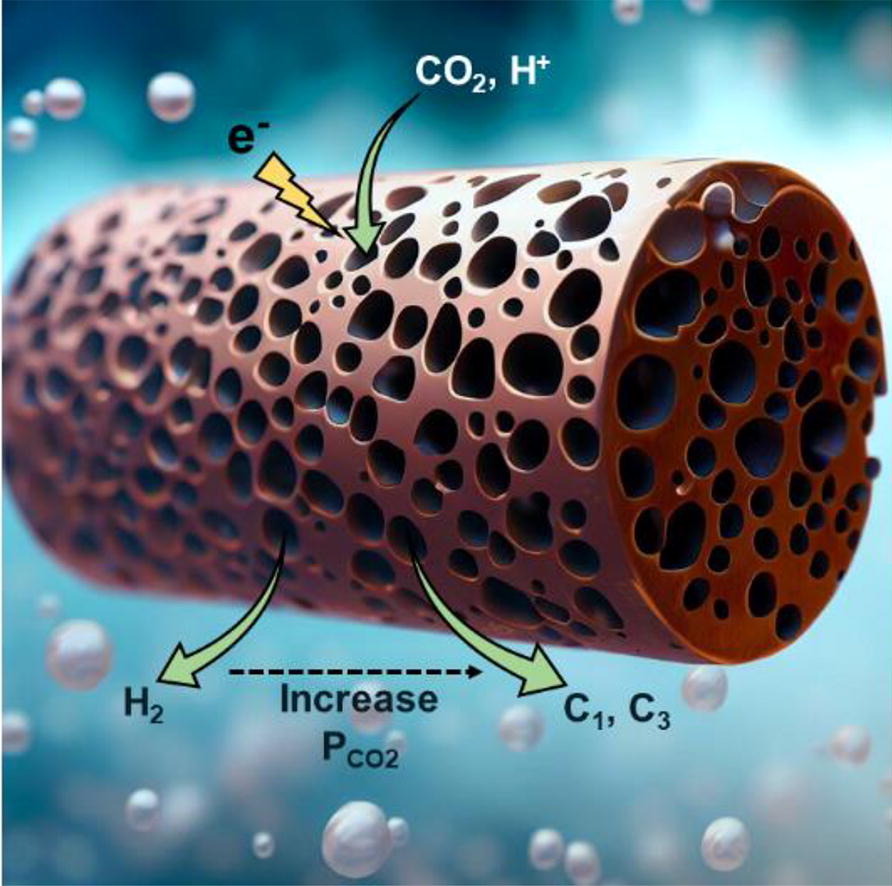
Electrochemical CO2 reduction is a promising way of closing the carbon cycle while synthesizing useful commodity chemicals and fuels. One of the possible routes to scale up the process is CO2 reduction at elevated pressure, as this is a way to increase the concentration of poorly soluble CO2 in aqueous systems. Yet, not many studies focus on this route, owing to the inherent challenges with high-pressure systems, such as leaks, product quantification, and ease of operation. In this study, we use a high-pressure flow cell setup to investigate the impact of CO2 pressure on the electrochemical performance of a copper foam electrode for CO2 reduction within a pressure range of 1 to 25 bar. Our initial findings using a 0.5 M potassium bicarbonate (KHCO3) electrolyte show a consistent improvement in selectivity towards CO2 reduction products, with HCOOH being the dominant product. By conducting a systematic exploration of operating parameters including applied current density, applied CO2 pressure, cation effect, and electrolyte concentration, the selectivity towards formate (HCOOH) is optimized, achieving a remarkable 70 % faradaic efficiency (FE) under moderate conditions of 25 bar in a 0.5 M cesium bicarbonate (CsHCO3) electrolyte. Additionally, we report the synthesis of isopropanol with a FE of 11 % at the 25 bar in 0.5 M KHCO3 which is the highest reported selectivity towards isopropanol on copper using a bicarbonate system.
Nandalal Girichandran, Saeed Saedy, Ruud Kortlever
-
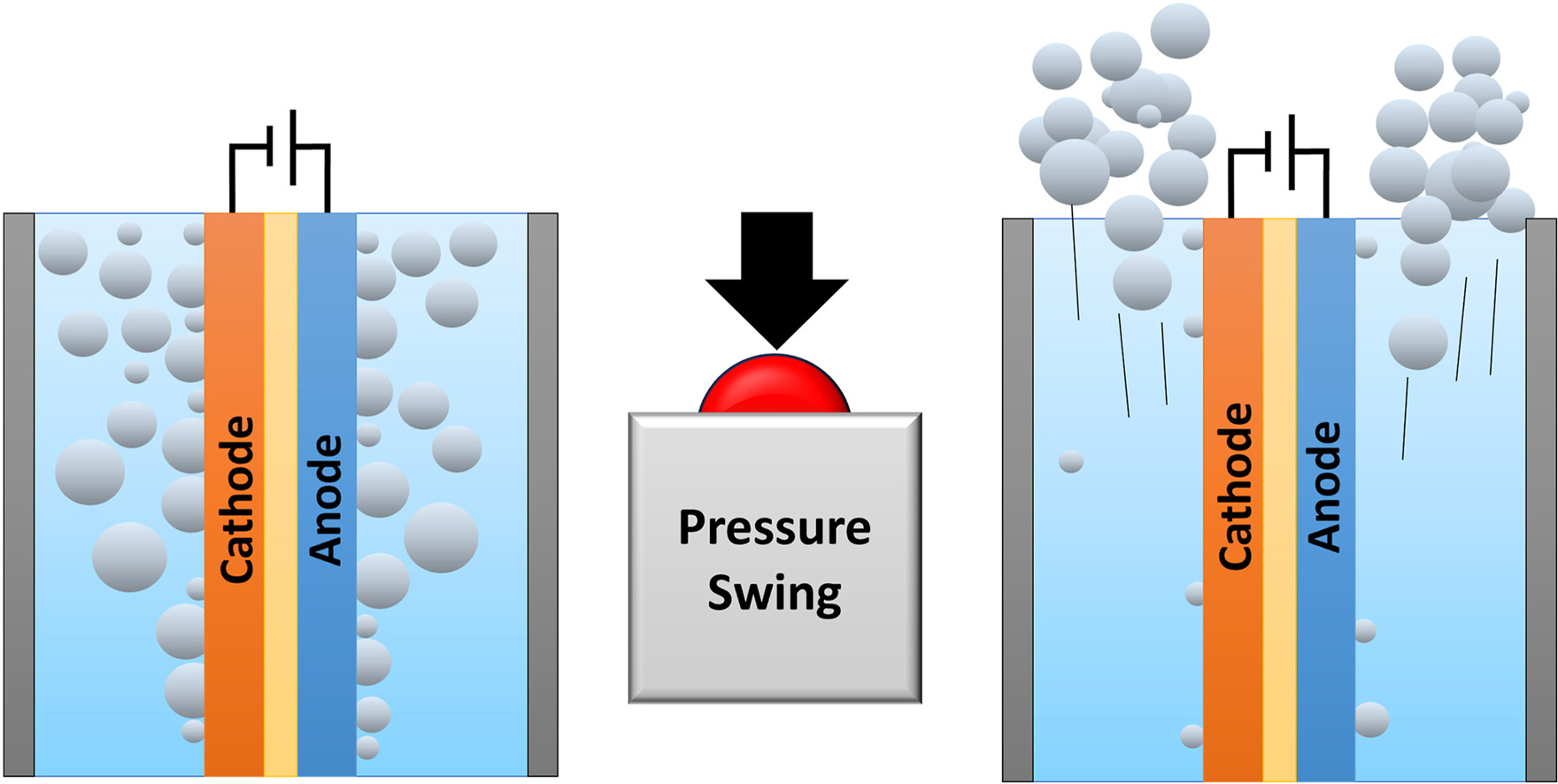
To make green hydrogen more economically attractive, the energy losses in alkaline electrolysis need to be minimized while operating at high current densities (1 A cm−2). At these current densities the ohmic resistance and gas bubbles effects contribute largely to the energy losses. To mitigate the gas bubbles losses, we demonstrate, for the first time, a pressure swing to remove gas bubbles in a zero-gap alkaline water electrolyzer. The pressure swing leverages the ideal gas law to increase the volume of gas in the system periodically, for a short duration (<2 s). This temporal volume increase effectively removes bubbles from the electrolyzer. We show that pressure swing can be used to measure the effect of bubbles on the ohmic resistance (RBubbles). Our results reveal that foam electrodes have a significantly larger RBubbles than perforated plate electrodes (1.8 Ω cm2 vs 0.3 Ω cm2). The time-averaged cell voltage reduces by 170 mV when applying pressure swings to an electrolyzer operating at 200 mA cm−2 in 1 M KOH with foam electrodes. The bubble resistance further depends on the electrolyte conductivity (inversely proportional) and is only moderately affected by operating pressure (25 % lower when increasing pressure amplitude from 1–2 to 1–5 bar). By implementing these findings in a model, we estimate that the pressure swing could reduce the cell voltage by ∼0.1 V for an electrolyzer operating at industrial conditions (6 M KOH, 80 °C, 1 A cm−2) for foam electrodes. For perforated plate electrodes, however, the reduced cell voltage is lower and does not outweigh the additional compression energy.
-
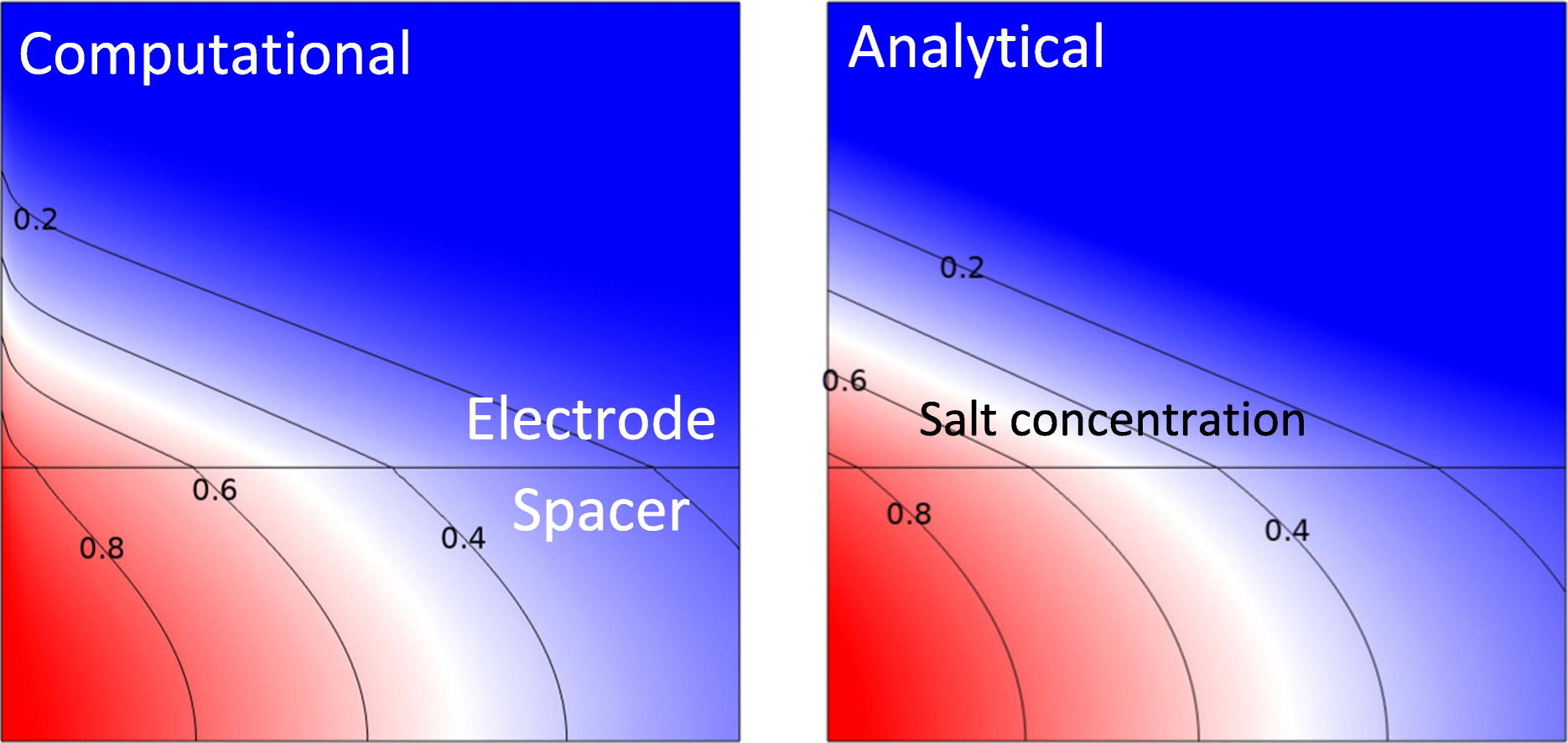
In flow-by capacitive deionization (CDI) brackish water flows between two electrodes that capacitively remove salt. We assume low inlet concentrations so “salt shocks” appear in the electrodes and the process becomes diffusion-limited. For unit charge efficiency, a simplified model is derived consisting of two coupled partial differential equations. We obtain approximations, and exact solutions in terms of the Lambert W function, for the salt concentration as a function of time and space and for the equilibrium charge-voltage relation. These surprisingly simple solutions compare well with the results from comprehensive two-dimensional simulations. Useful analytical expressions are obtained for optimal geometrical and operational parameters that maximize the productivity and minimize the specific energy losses. By making cells much thinner the productivity can be increased an order of magnitude compared to typical values in the literature. The optimal electrode is found to be roughly six times thinner than the spacer. The associated pressure drop is around 0.4 bar per 1 mM of inlet salt concentration, making our recommendations practically feasible only for relatively low concentrations. The obtained model and analytical expressions provide useful guidance to strongly improve the design process.
Haverkort, J. W., Sanderse, B., Padding, J. T., Blake, J. W.
-
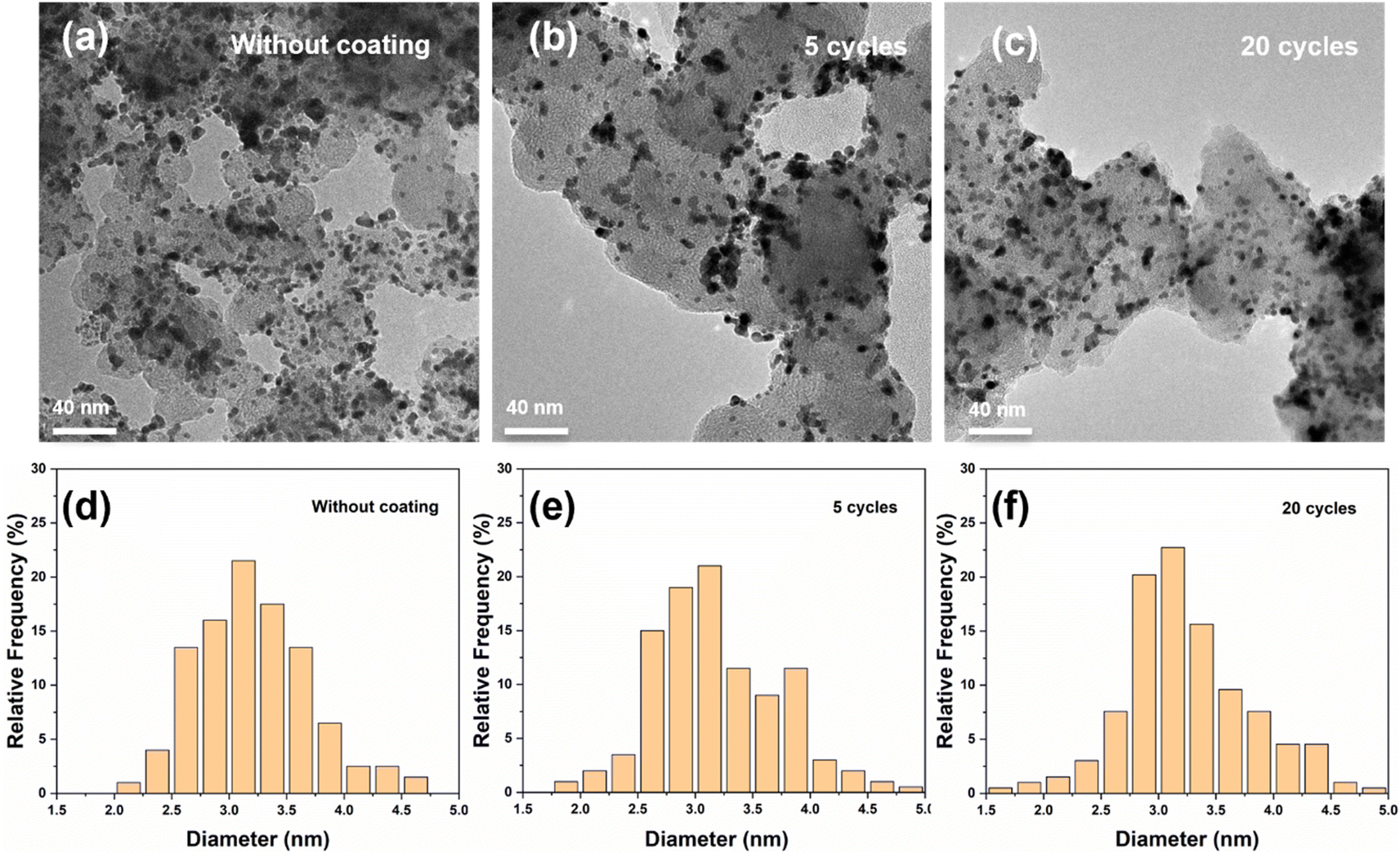
Extending the lifetime of electrocatalytic materials is a major challenge in electrocatalysis. Here, we employ atomic layer deposition (ALD) to coat the surface of carbon black supported platinum nanoparticles (Pt/CB) with an ultra-thin layer of silicon dioxide (SiO2) to prevent deactivation of the catalyst during H2 evolution. Our results show that after an accelerated durability test (ADT) the current density at −0.2 V vs. reversible hydrogen electrode (RHE) of the unprotected Pt/CB catalyst was reduced by 34%. By contrast, after coating the Pt/CB catalyst with 2 SiO2 ALD cycles, the current density at the same potential was reduced by 7% after the ADT procedure, whereas when the Pt/CB sample was coated with 5 SiO2 ALD cycles, the current density was reduced by only 2% after the ADT. Characterization of the Pt particles after electrochemical testing shows that the average particle size of the uncoated Pt/CB catalyst increases by roughly 16% after the ADT, whereas it only increases by 3% for the Pt/CB catalyst coated with 5 cycles of SiO2 ALD. In addition, the coating also strongly reduces the detachment of Pt nanoparticles, as shown by a strong decrease in the Pt concentration in the electrolyte after the ADT. However, 20 cycles of SiO2 ALD coating results in an over-thick coating that has an inhibitory effect on the catalytic activity. In summary, we demonstrate that only a few cycles of SiO2 ALD can strongly improve the stability of Pt catalyst for the hydrogen evolution reaction.
Ming Li, Saeed Saedy, Shilong Fu, Teise Stellema, Ruud Kortlever and J. Ruud van Ommen
-
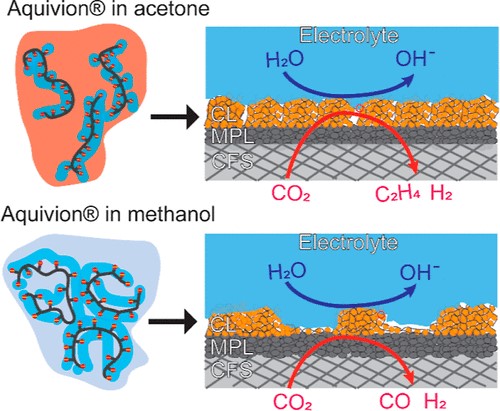
To explore the effects of solvent–ionomer interactions in catalyst inks on the structure and performance of Cu catalyst layers (CLs) for CO2 electrolysis, we used a “like for like” rationale to select acetone and methanol as dispersion solvents with a distinct affinity for the ionomer backbone or sulfonated ionic heads, respectively, of the perfluorinated sulfonic acid (PFSA) ionomer Aquivion. First, we characterized the morphology and wettability of Aquivion films drop-cast from acetone- and methanol-based inks on flat Cu foils and glassy carbons. On a flat surface, the ionomer films cast from the Aquivion and acetone mixture were more continuous and hydrophobic than films cast from methanol-based inks. Our study’s second stage compared the performance of Cu nanoparticle CLs prepared with acetone and methanol on gas diffusion electrodes (GDEs) in a flow cell electrolyzer. The effects of the ionomer–solvent interaction led to a more uniform and flooding-tolerant GDE when acetone was the dispersion solvent (acetone-CL) than when we used methanol (methanol-CL). As a result, acetone-CL yielded a higher selectivity for CO2 electrolysis to C2+ products at high current density, up to 25% greater than methanol-CL at 500 mA cm–2. Ethylene was the primary product for both CLs, with a Faradaic efficiency for ethylene of 47.4 ± 4.0% on the acetone-CL and that of 37.6 ± 5.5% on the methanol-CL at a current density of 300 mA cm–2. We attribute the enhanced C2+ selectivity of the acetone-CL to this electrode’s better resistance to electrolyte flooding, with zero seepage observed at tested current densities. Our findings reveal the critical role of solvent–ionomer interaction in determining the film structure and hydrophobicity, providing new insights into the CL design for enhanced multicarbon production in high current densities in CO2 electrolysis processes.
Y Wu, T Duignan, M Li, H Cartmill, I Maglaya, T Burdyny, G Wang, TE Rufford
-
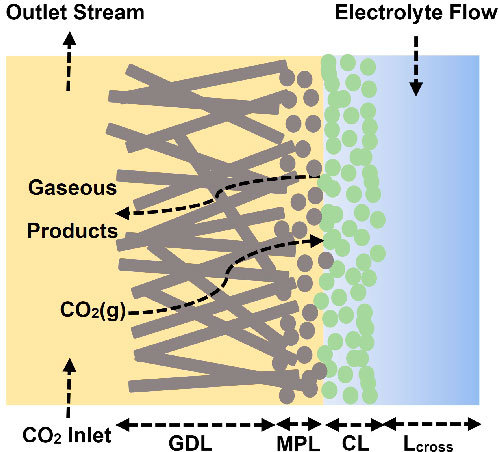
Electrochemical reduction of CO2 presents an attractive way to store renewable energy in chemical bonds in a potentially carbon-neutral way. However, the available electrolyzers suffer from intrinsic problems, like flooding and salt accumulation, that must be overcome to industrialize the technology. To mitigate flooding and salt precipitation issues, researchers have used super-hydrophobic electrodes based on either expanded polytetrafluoroethylene (ePTFE) gas-diffusion layers (GDL’s), or carbon-based GDL’s with added PTFE. While the PTFE backbone is highly resistant to flooding, the non-conductive nature of PTFE means that without additional current collection the catalyst layer itself is responsible for electron-dispersion, which penalizes system efficiency and stability. In this work, we present operando results that illustrate that the current distribution and electrical potential distribution is far from a uniform distribution in thin catalyst layers (~50 nm) deposited onto ePTFE GDL’s. We then compare the effects of thicker catalyst layers (~500 nm) and a newly developed non-invasive current collector (NICC). The NICC can maintain more uniform current distributions with 10-fold thinner catalyst layers while improving stability towards ethylene (≥ 30%) by approximately two-fold.
HP Iglesias van Montfort, M Li, E Irtem, M Abdinejad, Y Wu, S Pal, M Sassenburg, D Ripepi, J Biemolt, S Subramanian, T Rufford, T Burdyny
-
The electrochemical reduction of CO2 is increasingly seen as a viable means of producing carbon-based fuels and feedstocks due to the rapid advancement of cost-linked performance metrics within the past decade. These rapid advancements have also uncovered many fundamental and applied challenges (e.g., salt formation, CO2 utilization), which researchers have been systematically overcoming through various ingenuities at the catalyst, configuration, and operational levels. (1,2) Consequently, as the technology pushes further into the unknown and closer to commercially interesting performance metrics, the design, assembly, and operation of lab-scale CO2 electrolyzers must be regimented. In fact, such regulation is now necessary just to achieve the relevant baseline data needed to demonstrate new performance advancements. While many research studies report their experimental cells and systems used to generate their novel results, few provide an extensive overview, protocol, and system diagram that allows new researchers to reconstruct the entirety of the electrolysis system. Groups or new researchers entering the CO2 electrolysis research field must then either design systems themselves or incorporate pieces of information from a wide variety of sources.
HP Iglesias van Montfort, S Subramanian, E Irtem, M Sassenburg, M Li, J Kok, J Middelkoop, T Burdyny

-
Electrochemical CO2 capture is promising for closing the carbon cycle but needs technological advances. In a recent issue of Nature Energy, a novel chemistry for electrochemical CO2 capture is presented, demonstrating low energy consumption and high purity with virtually no degradation. This finally allows competition with amine-based capture technology.
David A. Vermaas, Ruud Kortlever
-
Most research into electrochemical CO2 conversion focusses on improving electrode materials, but neglects the role of the electrolyte. We show the buffer influence on the selectivity of a bimetallic gold–palladium electrode in an effort to elucidate observed inconsistencies between different studies. While hydrocarbons are produced in the phosphate buffer, they remain absent in the bicarbonate buffer, showing that the electrolyte choice plays a crucial role in the selectivity of the electrode.
Daniël van den Berg , Boaz Izelaar , Shilong Fu and Ruud Kortlever
This publication also appeared ont he cover of the Journal Catalysis, Science & Technology

-
The local conditions inside a gas diffusion electrode (GDE) pore, especially in the electrical double layer (EDL) region, influence the charge transfer reactions and the selectivity of desired CO2ER products. Most GDE computational models ignore the EDL or are limited in their applicability at high potentials. In this work, we present a continuum model to describe the local environment inside a catalytic pore at varying potentials, electrolyte concentrations and pore diameters. The systems studied in this work are based on an Ag catalyst in contact with KHCO3 solution. Our study shows that steric effects dominate the local environment at high cathodic potentials (≪−25 mV vs pzc at the OHP), leading to a radial drop of CO2 concentration. We also observe a drop in pH value within 1 nm of the reaction plane due to electrostatic repulsion and attraction of OH− and H+ ions, respectively. We studied the influence of pore radii (1–10 nm) on electric field and concentrations. Pores with a radius smaller than 5 nm show a higher mean potential, which lowers the mean CO2 concentration. Pores with a favourable local environment can be designed by regulating the ratio between the pore radius and Debye length.
Esaar N. Butt, Johan T. Padding and Remco Hartkamp
-
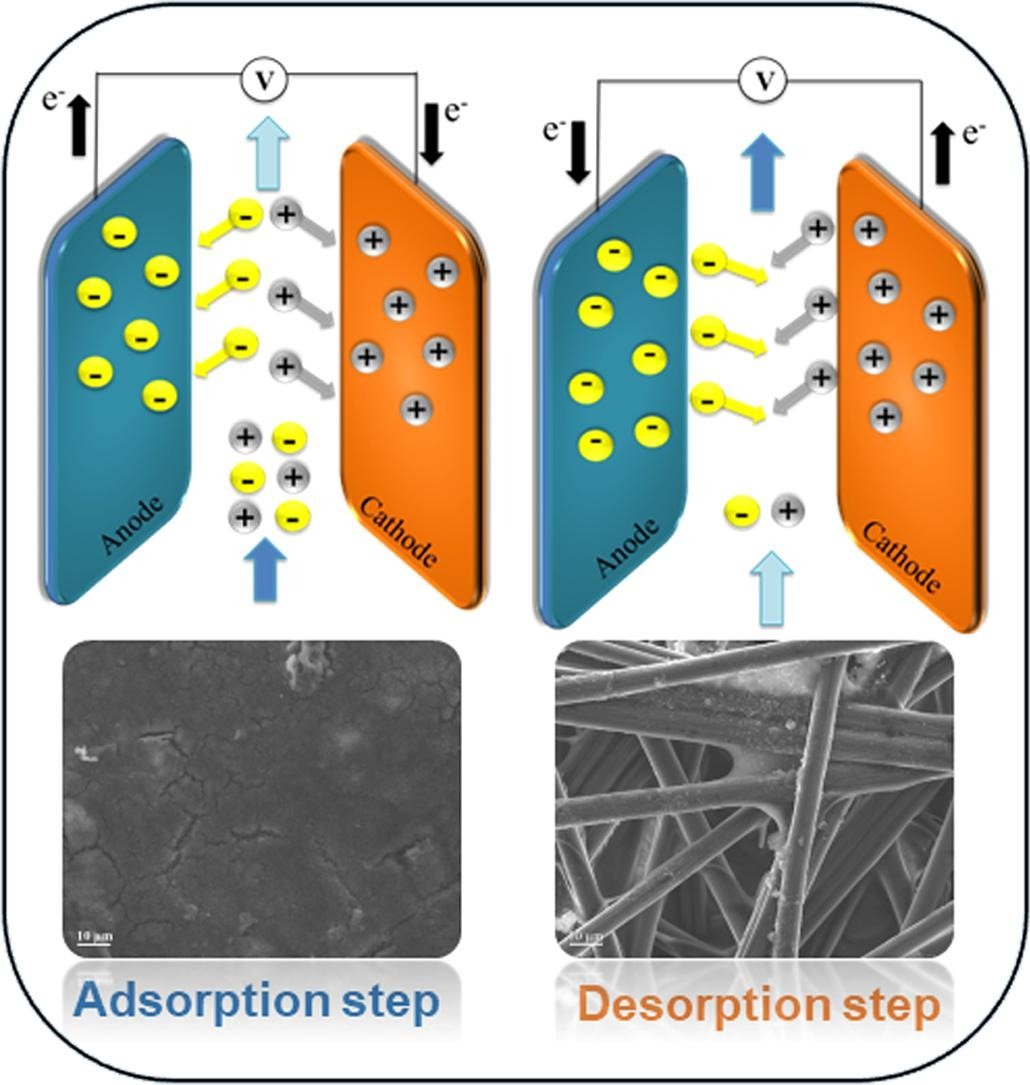
Selective ion separation is a fundamental challenge with applications ranging from the manufacturing of pharmaceuticals & industrial salts to water desalination. In particular, the separation of formate, a primary product of electrochemical carbon dioxide reduction, has attracted attention not only to reduce carbon emissions and energy costs but to provide new routes to value-added chemicals. In the present study, selective formate separation from an aqueous solution is demonstrated using an electrochemical flow cell with symmetric redox-active polyvinyl ferrocene electrodes. An electrosorption system equipped with an electrosorption cell, inline conductivity, and pH sensors was constructed to provide real-time measurements of the formate adsorption performance in continuous flow mode while varying operating conditions such as the flow rate, cell voltage, and electrolyte concentration. These parameters were optimized using a Box–Behnken experimental design to improve the formate adsorption selectivity. The flow cell results showed a selectivity higher than 6.0 toward the removal of formate in an electrolyte containing a 30-fold excess of perchlorate under optimal operation conditions (i.e., 0.5 mL/min flow rate, 1.0 V, and 15 mM electrolyte concentration). The performance of the flow cell was also tested using a solution that contained different liquid CO2 reduction products, and formate separation was achieved. The results suggest that the proper design of the electrochemical cell and efficient operation of the flow platform pave the way for scaling up the technology for selective formate separation.
Sevgi Polat, Ruud Kortlever, Hüseyin Burak Eral
-
Electrochemical carbon dioxide (CO2) reduction is a promising route to convert intermittent renewable energy into fuels and valuable chemical products. Separation of CO2 reduction products by ion-selective electrochemical technology may play a decisive role in the pursuit of commercially viable CO2 reduction processes. Selective separation of formate, one of the main CO2 reduction products, is assessed in the present study in an electrochemical flow cell with symmetric redox-active polyvinyl ferrocene (PVF) functionalized graphene oxide (GO) electrodes. First, experimental parameters such as the PVF/GO ratio, sonication time, and ultrasonic amplitude, were optimized in the electrode preparation process to improve the formate adsorption efficiency on a lab scale (1 × 2 cm electrodes) under static conditions. The electrochemical and morphological characteristics of the electrodes were investigated by cyclic voltammetry and scanning electron microscopy. To demonstrate continuous-flow operation, an electrosorption flow cell (8 × 8 cm) providing inline measurements was constructed. The flow cell results showed selectivity at > 5.5 toward the removal of formate from an electrolyte containing perchlorate at an excess of 30 times the normal value. The performance of the electrosorption cell was also tested using a mixture of methanol, ethanol, formate, and acetaldehyde produced in a CO2 reduction electrolyzer. In this demonstration, formate separation was achieved with a selectivity of > 4.0. The results suggest that the optimized design of the electrochemical cell and operation conditions of the flow platform pave the way for scaling up selective formate separation with PVF/GO electrodes.
Sevgi Polat, Ruud Kortlever, Hüseyin Burak Eral
-
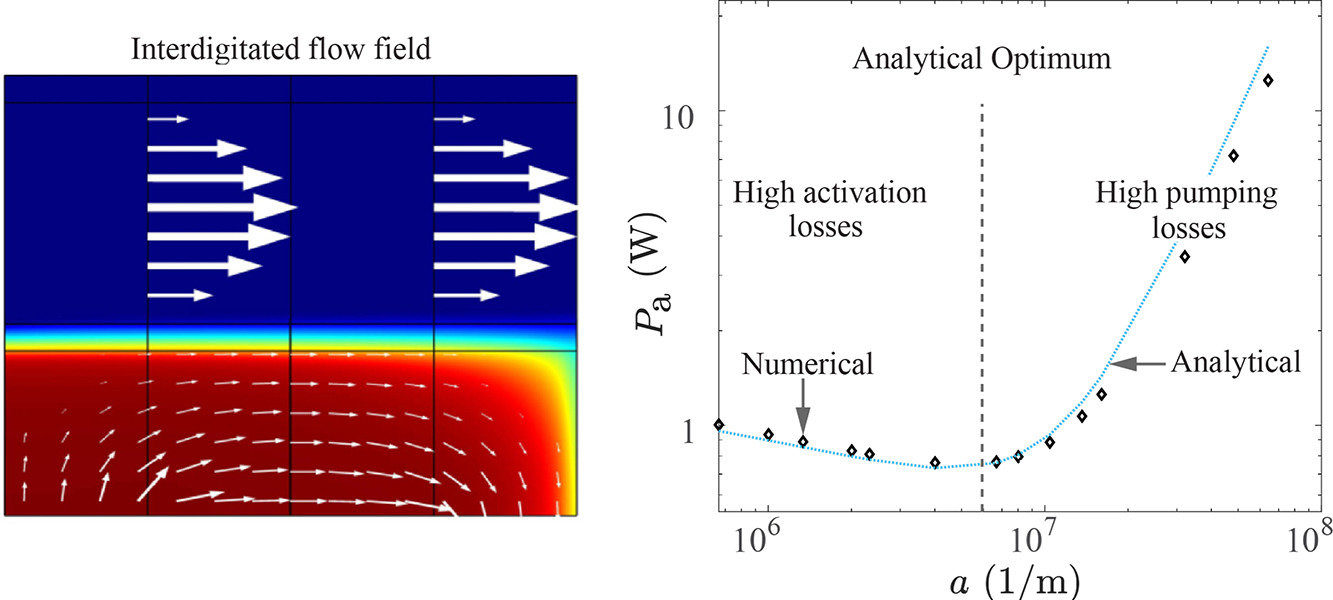
Microfluidic fuel cells, electrolyzers, and redox flow batteries utilize laminar flow channels to provide reactants, remove products and avoid their crossover. These devices often also employ porous flow-through electrodes as they offer a high surface area for the reaction and excellent mass transfer. The geometrical features of these electrodes and flow channels strongly influence energy efficiency. We derive explicit analytical relations for the optimal flow channel width and porous electrode volumetric surface area from the perspective of energy efficiency. These expressions are verified using a two-dimensional tertiary current distribution and porous electrode flow model in COMSOL and are shown to be able to predict optimal parameters in commonly used flow-through and interdigitated flow fields. The obtained analytical models can dramatically shorten modelling time and expedite the industrial design process. The optimal channel width and pore sizes we obtain, in the order of 100 microns and 1 micron respectively, are much smaller than those often used. This shows that there is a significant room for improvement of energy efficiency in flow cells that can sustain the resulting pressure drop.
A. Bhadra, J.W. Haverkort
-
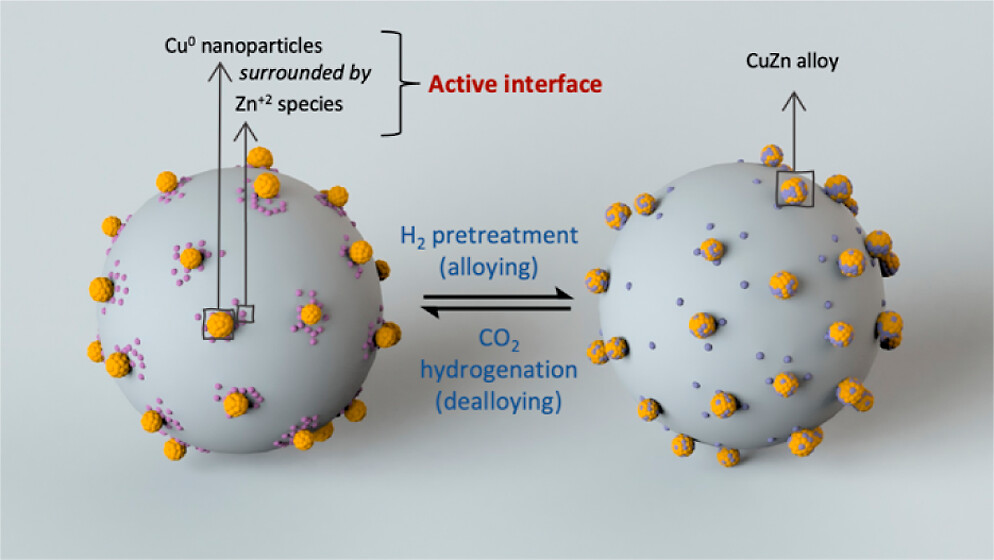
The direct synthesis of methanol via the hydrogenation of CO2, if performed efficiently and selectively, is potentially a powerful technology for CO2 mitigation. Here, we develop an active and selective Cu–Zn/SiO2 catalyst for the hydrogenation of CO2 by introducing copper and zinc onto dehydroxylated silica via surface organometallic chemistry and atomic layer deposition, respectively. At 230 °C and 25 bar, the optimized catalyst shows an intrinsic methanol formation rate of 4.3 g h–1 gCu–1 and selectivity to methanol of 83%, with a space-time yield of 0.073 g h–1 gcat–1 at a contact time of 0.06 s g mL–1. X-ray absorption spectroscopy at the Cu and Zn K-edges and X-ray photoelectron spectroscopy studies reveal that the CuZn alloy displays reactive metal support interactions; that is, it is stable under H2 atmosphere and unstable under conditions of CO2 hydrogenation, indicating that the dealloyed structure contains the sites promoting methanol synthesis. While solid-state nuclear magnetic resonance studies identify methoxy species as the main stable surface adsorbate, transient operando diffuse reflectance infrared Fourier transform spectroscopy indicates that μ-HCOO*(ZnOx) species that form on the Cu–Zn/SiO2 catalyst are hydrogenated to methanol faster than the μ-HCOO*(Cu) species that are found in the Zn-free Cu/SiO2 catalyst, supporting the role of Zn in providing a higher activity in the Cu–Zn system.
Hui Zhou, Scott R. Docherty, Nat Phongprueksathat, Zixuan Chen, Andrey V. Bukhtiyarov, Igor P. Prosvirin, Olga V. Safonova, Atsushi Urakawa, Christophe Copéret, Christoph R. Müller*, and Alexey Fedorov
-
Recently, carbon capture and reduction (CCR) technology has gained interest to directly convert CO2 to value-added products without requiring purification of CO2 and its subsequent transportation. CCR to methanol in one dual function material (DFM) poses mechanistic and kinetic challenges. To counteract this, a process combining Na/Al2O3 as a capture component and Cu/ZnO/Al2O3 (CZA) as methanol synthesis catalyst was developed to allow CCR to methanol. With a 5 vol% CO2 flow for capture and subsequent H2 stream combined with a temperature swing, a methanol selectivity of 26 % was achieved at 9 bar. Further investigation found that Na/Al2O3 significantly increased methanol yield, while a stacked configuration of Na/Al2O3 followed by CZA significantly outperformed a mixed configuration of the two catalysts. With further investigation of operation at higher pressure and surface mechanism, an effective CCR to methanol process using two affordable yet readily available catalysts can be realized.
Luca C. Wirner, Fumihiko Kosaka, Tomone Sasayama, Yanyong Liu, Atsushi Urakawa, Koji Kuramoto
-
Reaching our climate goals will require urgent advancements in the development of fossil-free technologies. Solid-oxide electrolysis (SOE) at high-temperature is a promising candidate for combining CO₂ utilization and renewable electricity use. Explorative techno-economic analyses are being performed to understand the full plant design requirements for integrated SOE systems. However, there is still a lack of understanding of the potential impact that the pre-treatment of CO₂ will have on the overall design and economics of a SOE-based system. To address this knowledge gap, as a first step, the process model of the pre-treatment units needed to purify CO₂ from a bioethanol plant is developed in Aspen Plus in the current work. Based on the preliminary results of this paper, the equipment costs mainly stem from the units related to the removal of sulfur (~65%) and alcohols (~32%). The energy costs are almost entirely related to the cryogenic distillation step required for the removal of non-condensable gases (~96%).
Josephine Vos, Andrea Ramirez, Mar Pérez-Fortes
-
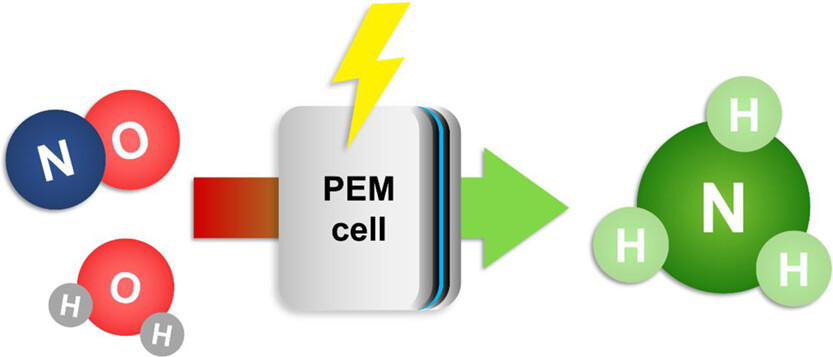
The continuous electrochemical NO reduction to ammonia in a PEM cell was investigated in this work. We used a ruthenium-based catalyst at the cathode and an iridium oxide catalyst at the anode. The highest ammonia faradaic efficiency was observed at 1.9 V cell voltage. Adjusting the NO flow allowed to achieve 97% NO conversion and 93% ammonia faradaic efficiency for a 5.2% NO/He feed. The ammonia yield was 0.51 mmol cm–2 h–1, among the highest reported to date with the advantage of continuous operation. Experiments with a low NO concentration feed of 983 ppm showed 98% conversion at 0 V vs pseudo-RHE. Achieving this performance under such mild conditions indicates the great potential of the PEM cells for NOx abatement applications and the production of valuable NH3.
Sorin Bunea, Manoj Coppens, and Atsushi Urakawa
-
Due to the heavy dependence on fossil-fuels as raw materials, the defossilization of feedstocks in the petrochemical industry represents a challenge. A large number of possible process routes that use alternative carbon sources (ACS) like CO2, biomass, and waste are being developed for the feedstock replacement. For instance, to produce ethylene, more than 40 ACS process routes were identified. These multiple options make the selection of the promising process route a complex task. By replacing feedstocks, a process can change significantly and the impacts related to these changes in a highly interconnected industrial cluster can create cascading effects due to system interdependencies. This work aims to understand the cascading impacts in carbon flows and prices of implementing an ACS production process in an ethylene cluster. The results show that PVC will be the highest impacted and defossilizing one value-chain can have cascading effect on other value-chains as observed for PET.
James Tonny Manalal, Mar Pérez-Fortes, Paola Ibarra Gonzalez, Andrea Ramirez Ramirez
-
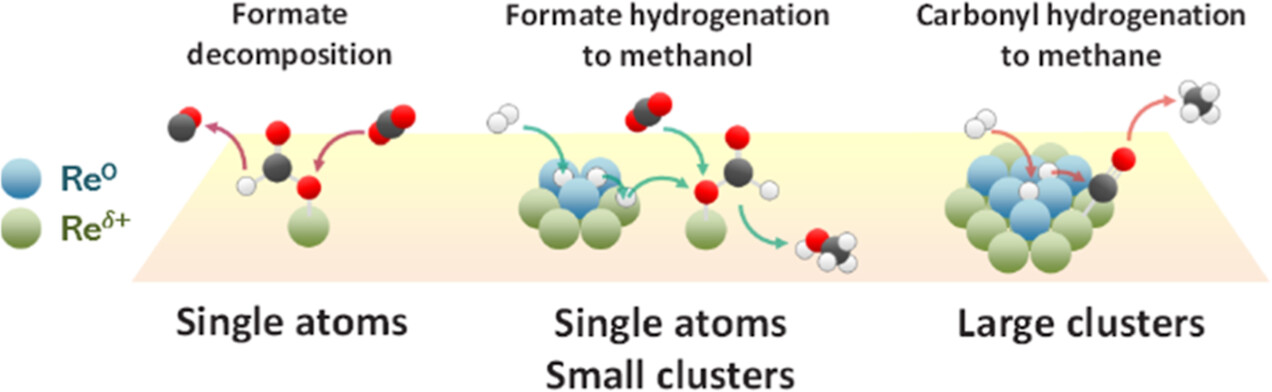
Low temperature and high pressure are thermodynamically more favorable conditions to achieve high conversion and high methanol selectivity in CO2 hydrogenation. However, low-temperature activity is generally very poor due to the sluggish kinetics, and thus, designing highly selective catalysts active below 200 °C is a great challenge in CO2-to-methanol conversion. Recently, Re/TiO2 has been reported as a promising catalyst. We show that Re/TiO2 is indeed more active in continuous and high-pressure (56 and 331 bar) operations at 125–200 °C compared to an industrial Cu/ZnO/Al2O3 catalyst, which suffers from the formation of methyl formate and its decomposition to carbon monoxide. At lower temperatures, precise understanding and control over the active surface intermediates are crucial to boosting conversion kinetics. This work aims at elucidating the nature of active sites and active species by means of in situ/operando X-ray absorption spectroscopy, Raman spectroscopy, ambient-pressure X-ray photoelectron spectroscopy (AP-XPS), and diffuse reflectance infrared Fourier transform spectroscopy (DRIFTS). Transient operando DRIFTS studies uncover the activation of CO2 to form active formate intermediates leading to methanol formation and also active rhenium carbonyl intermediates leading to methane over cationic Re single atoms characterized by rhenium tricarbonyl complexes. The transient techniques enable us to differentiate the active species from the spectator one on TiO2 support, such as less reactive formate originating from spillover and methoxy from methanol adsorption. The AP-XPS supports the fact that metallic Re species act as H2 activators, leading to H-spillover and importantly to hydrogenation of the active formate intermediate present over cationic Re species. The origin of the unique reactivity of Re/TiO2 was suggested as the coexistence of cationic highly dispersed Re including single atoms, driving the formation of monodentate formate, and metallic Re clusters in the vicinity, activating the hydrogenation of the formate to methanol.
Nat Phongprueksathat, Kah Wei Ting, Shinya Mine, Yuan Jing, Ryo Toyoshima, Hiroshi Kondoh, Ken-ichi Shimizu, Takashi Toyao, and Atsushi Urakawa
-
Electrochemical reduction of CO2 (CO2ER) is an emerging technology with the potential to limit the use of fossil-based feedstocks in the petrochemical industry by converting CO2 and renewable electricity into useful products such as syngas. Its successful deployment will depend not only on the technology's performance but also on its integration into the supply chain. In this work, a facility location model is used to gain insights regarding the capacity of CO2ER plants that produce syngas and the implications for the central/decentral placement of these CO2-based syngas plants. Different optimal configurations are examined in the model by changing the syngas transport costs. In this exploratory case, the results indicate that centralization is only an option when the syngas and CO2 transport costs are similar. When syngas transport is more expensive, decentralizing CO2-based syngas plants in the supply chain appears more feasible.
Thijmen Wiltink, Stijn Yska, Andrea Ramirez, Mar Pérez-Fortes
-
Using alternative carbon sources (ACS) to produce downstream derivatives (DDs) is a promising option for defossilising the chemical industry. However, the potential consequences of using ACS in interconnected petrochemical clusters are generally overlooked. This paper aims to develop a methodological approach for systematically analysing defossilisation impacts at the value chain level. For this, a single value chain for producing methyl-tert-butyl-ether (MTBE) was used as a case study. The individual components of the value chain were modelled in Aspen Plus v12. Both ACS- and fossil-based value chains were compared in terms of (i) changes in the structure of the value chain and (ii) the magnitude of the impacts. The results show that the defossilisation of a single value chain causes additional impacts at the cluster level.
Inna Stepchuk, Mar Pérez-Fortes, Andrea Ramírez Ramírez
-
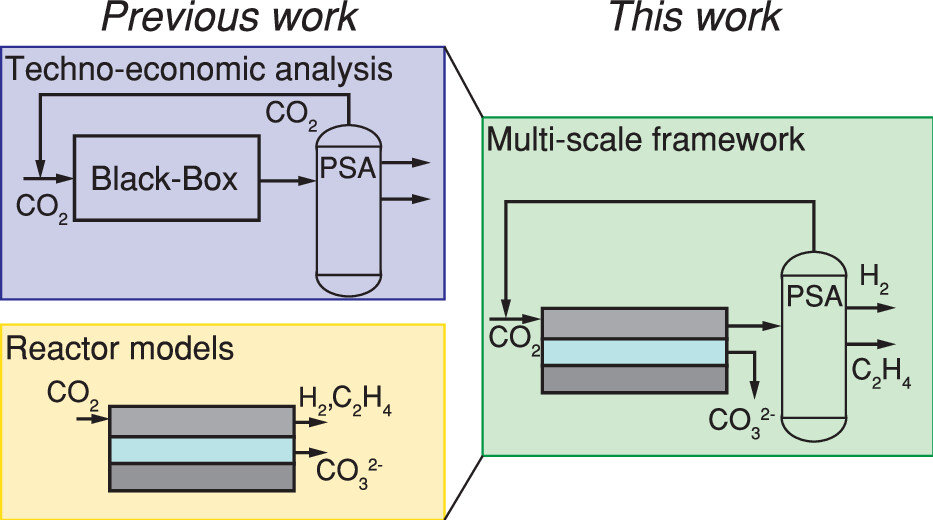
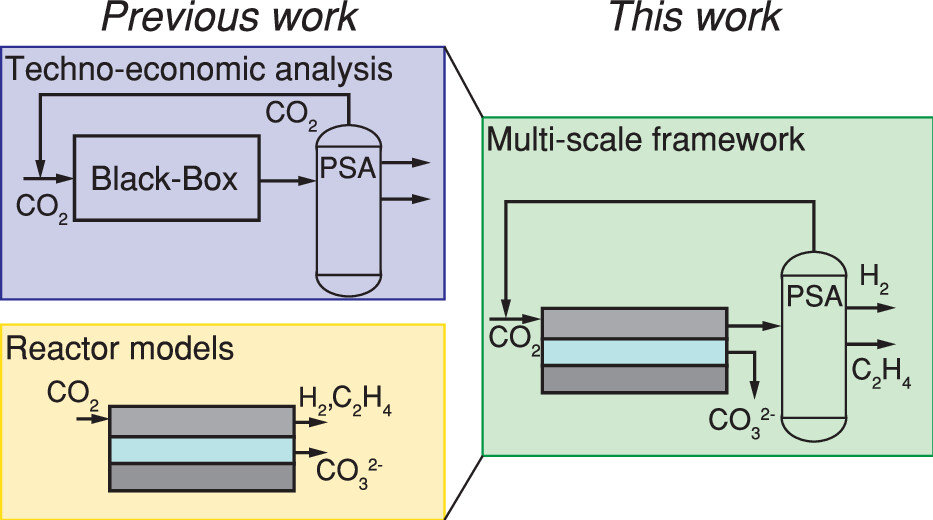
The production of base chemicals by electrochemical conversion of captured CO2 has the potential to close the carbon cycle, thereby contributing to a future energy transition. With the feasibility of low-temperature electrochemical CO2 conversion demonstrated at lab scale, research is shifting toward optimizing electrolyser design and operation for industrial applications, with target values based on techno-economic analysis. However, current techno-economic analyses often neglect experimentally reported interdependencies of key performance variables such as the current density, the faradaic efficiency, and the conversion. Aiming to understand the impact of these interdependencies on the economic outlook, we develop a model capturing mass transfer effects over the channel length for an alkaline, membrane electrolyser. Coupling the channel scale with the higher level process scale and embedding this multiscale model in an economic framework allows us to analyze the economic trade-off between the performance variables. Our analysis shows that the derived target values for the performance variables strongly depend on the interdependencies described in the channel scale model. Our analysis also suggests that economically optimal current densities can be as low as half of the previously reported benchmarks. More generally, our work highlights the need to move toward multiscale models, especially in the field of CO2 electrolysis, to effectively elucidate current bottlenecks in the quest toward economically compelling system designs.
Isabell Bagemihl, Lucas Cammann, Mar Pérez-Fortes, Volkert van Steijn, J. Ruud van Ommen
-
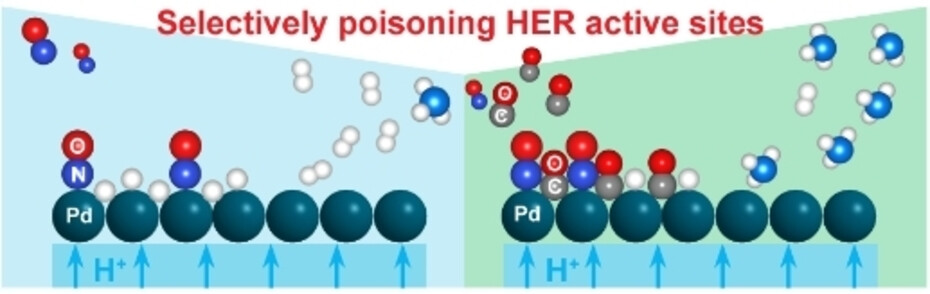
Direct electroreduction of nitric oxide offers a promising avenue to produce valuable chemicals, such as ammonia, which is an essential chemical to produce fertilizers. Direct ammonia synthesis from NO in a polymer electrolyte membrane (PEM) electrolyzer is advantageous for its continuous operation and excellent mass transport characteristics. However, at a high current density, the faradaic efficiency of NO electroreduction reaction is limited by the competing hydrogen evolution reaction (HER). Herein, we report a CO-mediated selective poisoning strategy to enhance the faradaic efficiency (FE) towards ammonia by suppressing the HER. In the presence of only NO at the cathode, Pt/C and Pd/C catalysts showed a lower FE towards NH3 than to H2 due to the dominating HER. Cu/C catalyst showed a 78 % FE towards NH3 at 2.0 V due to the stronger binding affinity to NO* compared to H*. By co-feeding CO, the FE of Cu/C catalyst towards NH3 was improved by 12 %. More strikingly, for Pd/C, the FE towards NH3 was enhanced by 95 % with CO co-feeding, by effectively suppressing HER. This is attributed to the change of the favorable surface coverage resulting from the selective and competitive binding of CO* to H* binding sites, thereby improving NH3 selectivity.
Min Li, Jarco Verkuil, Dr. Sorin Bunea, Dr. Ruud Kortlever, Prof. Dr. Atsushi Urakawa
-
This review provides a comprehensive overview of the dynamics of low-temperature water electrolyzers and their influence on coupling the three major technologies, alkaline, Proton Exchange Membrane (PEM) and, Anion Exchange Membrane (AEM) with photovoltaic (PV) systems. Hydrogen technology is experiencing considerable interest as a way to accelerate the energy transition. With no associated CO2 emissions and fast response, water electrolyzers are an attractive option for producing green hydrogen on an industrial scale. This can be seen by the ambitious goals and large-scale projects being announced for hydrogen, especially with solar energy dedicated entirely to drive the process. The electrical response of water electrolyzers is extremely fast, making the slower variables, such as temperature and pressure, the limiting factors for variable operation typically associated with PV-powered electrolysis systems. The practical solar-to-hydrogen efficiency of these systems is in the range of 10% even with a very high coupling factor exceeding 99% for directly coupled systems. The solar-to-hydrogen efficiency can be boosted with a battery, potentially sacrificing the cost. The intermittency of solar irradiance, rather than its variability is the biggest challenge for PV-hydrogen systems regarding operation and degradation.
V.A. Martinez Lopez, H. Ziar, J.W. Haverkort, M. Zeman, O. Isabella
-
Ammonia (NH3) ranks among the largest bulk chemical products in the world, with an annual production of 178 million tons and an estimated annual market growth of 3–5% to meet the global demand for fertilizer in the agricultural sector due to an increasing world population. (1,2) The majority of NH3 is produced by the Haber–Bosch process, wherein elevated temperatures (300–500 °C) and pressures (200–300 bar) are required. (3) In addition, the current process has a major environmental impact (∼1% of the global greenhouse emissions), mostly due to the production of hydrogen by steam-methane reforming. (4) To meet the net-zero emissions goal by 2050, as established in the latest IPCC report, (5) ammonia must be produced via a sustainable pathway. (6) Direct electrocatalytic synthesis of ammonia from dinitrogen and water at mild conditions could potentially offer a carbon-free alternative, resilient to intermittent renewable energy generation. (7)
Boaz Izelaar, Davide Ripepi, Dylan D. van Noordenne, Peter Jungbacker, Ruud Kortlever*, and Fokko M. Mulder
-
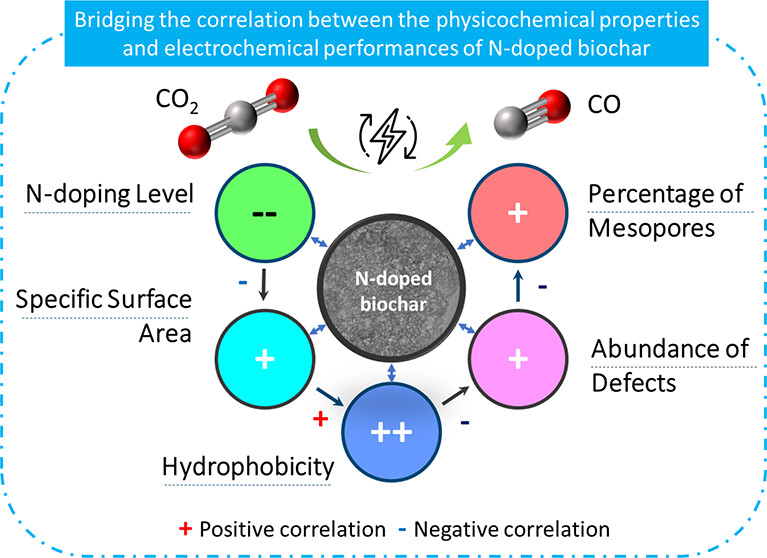
Nitrogen-doped (N-doped) carbon catalysts have been widely studied for electrochemical CO2 reduction to CO. However, the correlation between the physicochemical properties of N-doped carbon catalysts and their electrocatalytic performance for the CO2RR is still unclear. Herein, a series of N-doped biochar catalysts with different physicochemical properties were synthesized by tuning the carbonization temperature and N-doping level and used for the CO2RR to analyze the structure–performance relationship. The prepared catalysts exhibited massive differences in maximum faradaic efficiency to CO from 26.8 to 94.9% at around −0.8 to −0.9 V vs RHE. In addition, we find that simply increasing the specific surface area and N-doping level of the catalysts does not effectively improve the catalytic performance for the CO2RR. A multivariate correlation analysis reveals a negative correlation between the N-doping content and the electrochemical performance. The porous structural properties exhibit a positive correlation to the FECO but almost no correlation to jCO. Interestingly, improving the degree of graphitization, surface hydrophobicity, the abundance of defects, and optimizing the porosity of the N-doped biochar catalyst can efficiently enhance the catalytic performance for the CO2RR. We conclude that comprehensively analyzing the synergistic effect of various properties of N-doped biochar is critical to reveal structure–activity relationships.
Shilong Fu, Ming Li, Wiebren de Jong, and Ruud Kortlever
-
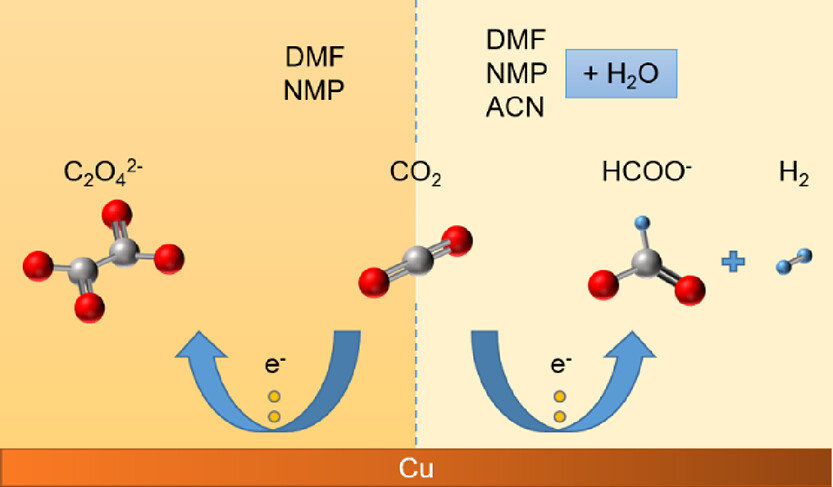
Aqueous electrolytes used in CO2 electroreduction typically have a CO2 solubility of around 34 mM under ambient conditions, contributing to mass transfer limitations in the system. Non-aqueous electrolytes exhibit higher CO2 solubility (by 5–8-fold) and also provide possibilities to suppress the undesired hydrogen evolution reaction (HER). On the other hand, a proton donor is needed to produce many of the products commonly obtained with aqueous electrolytes. This work investigates the electrochemical CO2 reduction performance of copper in non-aqueous electrolytes based on dimethylformamide (DMF), n-methyl-2-pyrrolidone (NMP), and acetonitrile (ACN). The main objective is to analyze whether non-aqueous electrolytes are a viable alternative to aqueous electrolytes for hydrocarbon production. Additionally, the effects of aqueous/non-aqueous anolytes, membrane, and the selection of a potential window on the electrochemical CO2 reduction performance are addressed in this study. Experiments with pure DMF and NMP mainly produced oxalate with a faradaic efficiency (FE) reaching >80%; however, pure ACN mainly produced hydrogen and formate due to the presence of more residual water in the system. Addition of 5% (v/v) water to the non-aqueous electrolytes resulted in increased HER and formate production with negligible hydrocarbon production. Hence, we conclude that aqueous electrolytes remain a better choice for the production of hydrocarbons and alcohols on a copper electrode, while organic electrolytes based on DMF and NMP can be used to obtain a high selectivity toward oxalate and formate.
Asvin Sajeev Kumar, Marilia Pupo, Kostadin V. Petrov, Mahinder Ramdin, J. Ruud van Ommen, Wiebren de Jong, and Ruud Kortlever
-
Immobilizing molecular catalysts on electrodes is vital for electrochemical applications. However, creating robust electrode-catalyst interactions while maintaining good catalytic performance and rapid electron transfer is challenging. Here, without introducing any foreign elements, we show a bottom-up synthetic approach of constructing the conjugated C−C bond between the commercial Vulcan carbon electrode and an organometallic catalyst. Characterization results from FTIR, XPS, aberration-corrected TEM and EPR confirmed the successful and uniform heterogenization of the complex. The synthesized Vulcan-LN4−Co catalyst is highly active and selective in the oxygen reduction reaction in neutral media, showing an 80 % hydrogen peroxide selectivity and a 0.72 V (vs. RHE) onset potential which significantly outperformed the homogenous counterpart. Based on single-crystal XRD and NMR data, we built a model for density functional theory calculations which showed a nearly optimal binding energy for the *OOH intermediate. Our results show that the direct conjugated C−C bonding is an effective approach for heterogenizing molecular catalysts on carbon, opening new opportunities for employing molecular catalysts in electrochemical applications.
Dr. Jasper Biemolt, Eva J. Meeus, Felix J. de Zwart, Jeen de Graaf, Petrus C. M. Laan, Prof. Dr. Bas de Bruin, Dr. Thomas Burdyny, Prof. Dr. Gadi Rothenberg, Prof. Dr. Ning Yan
-
The production of base chemicals by electrochemical conversion of captured CO2 has the potential to close the carbon cycle, thereby contributing to a future energy transition. With the feasibility of low-temperature electrochemical CO2 conversion demonstrated at lab scale, research is shifting toward optimizing electrolyser design and operation for industrial applications, with target values based on techno-economic analysis. However, current techno-economic analyses often neglect experimentally reported interdependencies of key performance variables such as the current density, the faradaic efficiency, and the conversion. Aiming to understand the impact of these interdependencies on the economic outlook, we develop a model capturing mass transfer effects over the channel length for an alkaline, membrane electrolyser. Coupling the channel scale with the higher level process scale and embedding this multiscale model in an economic framework allows us to analyze the economic trade-off between the performance variables. Our analysis shows that the derived target values for the performance variables strongly depend on the interdependencies described in the channel scale model. Our analysis also suggests that economically optimal current densities can be as low as half of the previously reported benchmarks. More generally, our work highlights the need to move toward multiscale models, especially in the field of CO2 electrolysis, to effectively elucidate current bottlenecks in the quest toward economically compelling system designs.
Isabell Bagemihl, Lucas Cammann, Mar Pérez-Fortes, Volkert van Steijn, and J. Ruud van Ommen
-
Yannai Kashtan and his colleagues propose that universities should sever research links with fossil-fuel companies (Nature 612, 404; 2022). We argue that collaboration in some areas is essential to ensure a timely transition to clean energy, given the substantial funding needed to quickly develop and scale up green technologies.
A transition to low-emitting processes will be slow and expensive for some industries. In the chemical industry, for instance, around 200 million tonnes of ethylene are produced annually. A newly developed sustainable process that produces 25,000 tonnes of ethylene per year would need 30 years to reach this output at a compound annual growth rate of 35%. Only companies that are already embedded into supply chains and the global energy infrastructure can reorient such mega-chemical production routes in a reasonable timeframe.
Universities need immense amounts of funding to train researchers, advance new technologies and develop prototypes. And industry understands the challenges that need to be addressed for successful upscaling. Working together will therefore benefit society, provided that the collaboration promotes a zero-emission energy system.
Thomas Burdyny and Bernard Dam
-
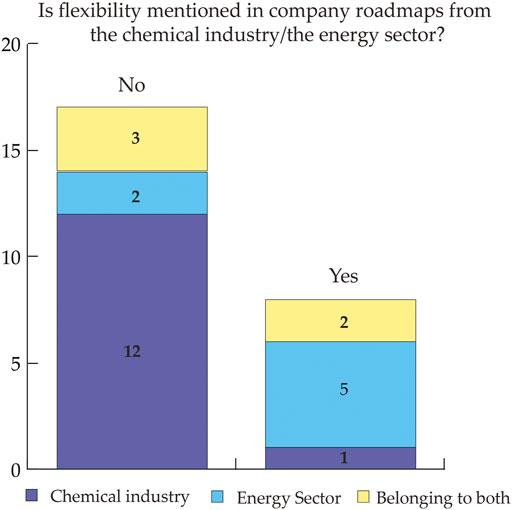
Electrification of processes and utilities is considered a promising option towards the reduction of greenhouse gas emissions from the chemical industry. Therefore, electricity demand is expected to increase steeply. Since the sources of future low-carbon electricity are variable in nature, there is a need for strategies to match availability and demand. Literature identified the flexibility of chemical processes as one promising strategy to address variability. This study aims to provide insights into how stakeholders from the power sector and the chemical industry consider flexibility in chemical processes and to identify key benefits and bottlenecks. For this article, we combined a review of peer-reviewed and grey literature with stakeholder interviews to map and describe the state of the art of flexible chemicals production, and to identify requirements for further research. The main drivers to investigate the flexibility potential are first, the contribution to energy system reliability, and second, potential cost savings for the industry. Main limitations are considered to be first, the uncertain economic performance of flexible processes due to investment costs, reduced production and uncertain revenues from flexible operation, and second, the complexity of the implementation of flexibility.
Svenja Bielefeld, Miloš Cvetković, Andrea Ramírez
-
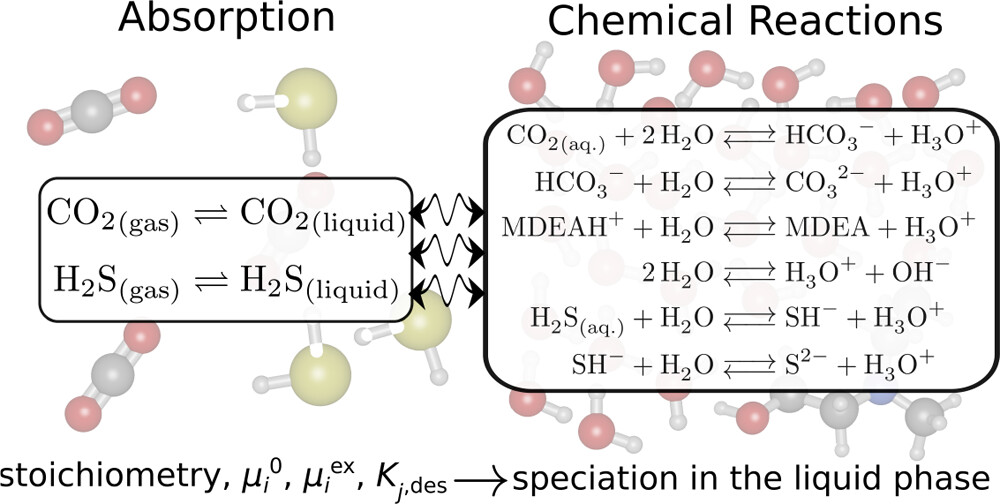
We developed an open-source chemical reaction equilibrium solver in Python (CASpy, github.com/omoultosEthTuDelft/CASpy) to compute the concentration of species in any reactive liquid-phase absorption system. We derived an expression for a mole fraction-based equilibrium constant as a function of excess chemical potential, standard ideal gas chemical potential, temperature, and volume. As a case study, we computed the CO2 absorption isotherm and speciation in a 23 wt % N-methyldiethanolamine (MDEA)/water solution at 313.15 K, and compared the results with available data from the literature. The results show that the computed CO2 isotherms and speciations are in excellent agreement with experimental data, demonstrating the accuracy and the precision of our solver. The binary absorptions of CO2 and H2S in 50 wt % MDEA/water solutions at 323.15 K were computed and compared with available data from the literature. The computed CO2 isotherms showed good agreement with other modeling studies from the literature while the computed H2S isotherms did not agree well with experimental data. The experimental equilibrium constants used as an input were not adjusted for H2S/CO2/MDEA/water systems and need to be adjusted for this system. Using free energy calculations with two different force fields (GAFF and OPLS-AA) and quantum chemistry calculations, we computed the equilibrium constant (K) of the protonated MDEA dissociation reaction. Despite the good agreement of the OPLS-AA force field (ln[K] = −24.91) with the experiments (ln[K] = −23.04), the computed CO2 pressures were significantly underestimated. We systematically investigated the limitations of computing CO2 absorption isotherms using free energy and quantum chemistry calculations and showed that the computed values of μiex are very sensitive to the point charges used in the simulations, which limits the predictive power of this method.
H. Mert Polat, Frédérick de Meyer, Céline Houriez, Othonas A. Moultos, and Thijs J. H. Vlugt
-
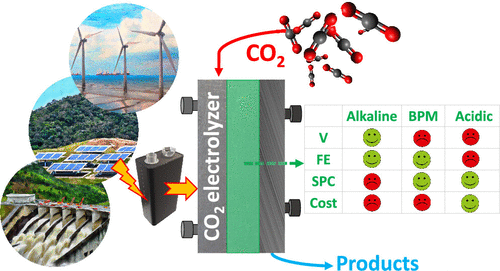
Electrochemical reduction of carbon dioxide (CO2) to useful products is an emerging power-to-X concept, which aims to produce chemicals and fuels with renewable electricity instead of fossil fuels. Depending on the catalyst, a range of chemicals can be produced from CO2 electrolysis at industrial-scale current densities, high Faraday efficiencies, and relatively low cell voltages. One of the main challenges for up-scaling the process is related to (bi)carbonate formation (carbonation), which is a consequence of performing the reaction in alkaline media to suppress the competing hydrogen evolution reaction. The parasitic reactions of CO2 with the alkaline electrolytes result in (bi)carbonate precipitation and flooding in gas diffusion electrodes, CO2 crossover to the anode, low carbon utilization efficiencies, electrolyte carbonation, pH-drift in time, and additional cost for CO2 and electrolyte recycling. We present a critical review of the causes, consequences, and possible solutions for the carbonation effect in CO2 electrolyzers. The mechanism of (bi)carbonate crossover in different cell configurations, its effect on the overall process design, and the economics of CO2 and electrolyte recovery are presented. The aim is to provide a better understanding of the (bi)carbonate problem and guide research directions to overcome the challenges related to low-temperature CO2 electrolysis in alkaline media.
Mahinder Ramdin, Othonas A. Moultos, Leo J. P. van den Broeke, Prasad Gonugunta, Peyman Taheri and Thijs J. H. Vlugt
-
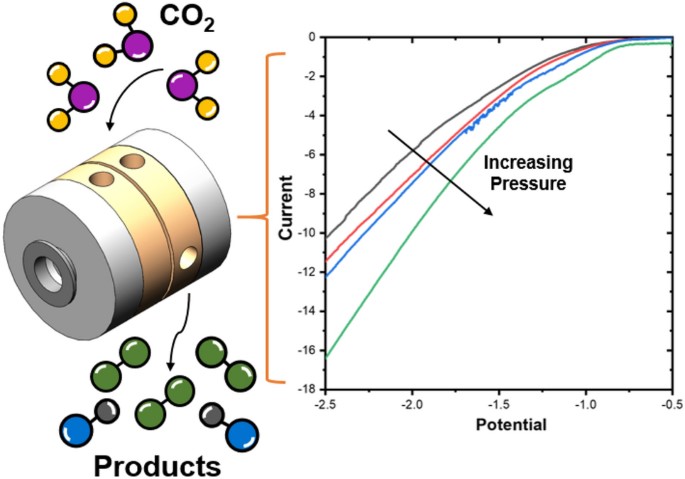
The electrochemical CO2 reduction reaction (CO2RR) has been proposed as a sustainable way of closing the carbon cycle while synthesizing useful commodity chemicals. One of the possible routes to scale up the process is the elevated pressure CO2RR, as this increases the concentration of the poorly soluble CO2 in aqueous systems. Yet, there are not many studies that focus on this route owing to the inherent challenges with high pressure systems. In this study, a novel high pressure flow cell setup has been designed and validated. The modular design uses a clamp system, which facilitates simple stacking of multiple cell parts while being capable of handling pressures up to 50 bar. The effects of CO2 pressure on the reaction were investigated on a gold (Au) foil cathode in a 0.1 M KHCO3 electrolyte. We successfully measured gaseous products produced during high pressure operation using an inline gas chromatograph. We find that the selectivity toward CO2 reduction products is enhanced while that of H2 evolution is suppressed as the pressure is increased from 2 to 30 bar. The reported setup provides a robust means to conduct high pressure electrolysis experiments in an easy and safe manner, making this technology more accessible to the electrochemical CO2RR community.
Andrew R. T. Morrison, Nandalal Girichandran, Quincy Wols & Ruud Kortlever
-
Understanding multiphase flow close to the electrode surface is crucial to the design of electrolyzers, such as alkaline water electrolyzers for the production of green hydrogen. Vertical electrodes develop a narrow gas plume near their surface. We apply the integral method to the mixture model. Considering both exponentially varying and step-function gas fraction profiles, we derive analytical relations for plume thickness, velocity profile, and gas fraction near the electrode as a function of height and current density. We verify these analytical relations with the numerical solutions obtained using two-dimensional mixture model simulations. We find that for low gas fractions, the plume thickness decreases with an increase in current density for an exponentially varying gas fraction profile. In contrast, the plume thickness increases with increasing current density at high gas fractions for an approximately step-function-shaped gas fraction profile, in agreement with experiments from the literature.
A. Rajora, J.W. Haverkort
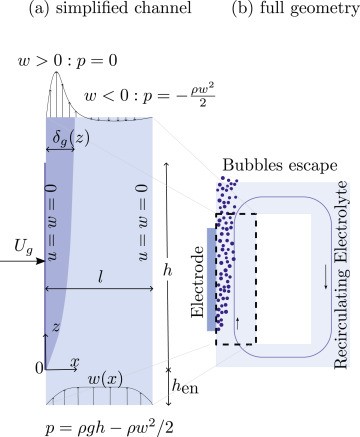
-
Formic acid production from CO2 allows the reduction of carbon dioxide emissions while synthesizing a product with a wide range of applications. CO2 hydrogenation is challenging due to the cost of transition metal catalysts and the toxicity of the transition elements. In this work, the thermodynamic confinement effects of the metal–organic framework UiO-66 on the CO2 hydrogenation to formic acid were studied by force field-based molecular simulations. The confinement effects of UiO-66 and the metal–organic frameworks Cu-BTC, and IRMOF-1 were compared, to assess the impact of different pore size and metal centers on the production of HCOOH. Monte Carlo simulations in the grand-canonical ensemble were performed in the frameworks, using gas phase mole fractions of CO2, H2, and HCOOH at chemical equilibrium, obtained from Continuous Fractional Component Monte Carlo simulations in the Reaction Ensemble. The adsorption isobars of the components in metal–organic frameworks were computed at 298.15 – 800 K, 1 – 60 bar. The enhancement of HCOOH production due to preferential adsorption of HCOOH in metal–organic frameworks was calculated for all studied conditions. UiO-66, Cu-BTC, and IRMOF-1 affect CO2 hydrogenation reaction, shifting the thermodynamical equilibrium toward HCOOH formation. The prevailing factor is the type of metal center in the metal–organic framework. The confinement effect of Cu-BTC turns out to exceed the enhancement caused by UiO-66, and IRMOF-1. The resulting mole fraction of HCOOH increased by ca. 2000 times compared to the gas phase at 298.15 K, 60 bar. Cu-BTC can be considered as an alternative to improve the production of HCOOH due to elimination of the high-cost temperature elevation, cost reduction of downstream processing methods, and comparable final concentration of HCOOH to the reported concentrations of formate obtained using transition metal catalysts.
Dominika O. Wasik, Ana Martín-Calvo, Juan José Gutiérrez-Sevillano, David Dubbeldam, Thijs J.H. Vlugt, Sofía Calero
-
Aqueous electrolytes are most commonly used for the CO2 reduction reaction (CO2RR), but suffer from a low CO2 solubility that limits the reaction. Electrochemical CO2 reduction in nonaqueous electrolytes can provide a solution, due to the higher CO2 solubility of organic solvent-based electrolytes. Herein, the product distribution of the electrochemical CO2 reduction on polycrystalline Cu in 0.7 m tetraethylammonium chloride in propylene carbonate with different water additions (0, 10, and 90 v%), and for different operating conditions (10, 25, 40, and 60 °C), is investigated. It is found that CO2 reduction on Cu in a propylene carbonate solution results in H2, CO, and formic acid formation only, even though Cu is known to produce C2+ products such as ethylene and ethanol in aqueous electrolytes. Increasing the operating temperature increases the CO2RR kinetics and shows an improvement in CO formation and decrease in H2 formation. However, increasing the operating temperature also increases water transport through the membrane, resulting in an increase of H2 formation over time when operating at 60 °C.
Iris Burgers, Elena Pérez-Gallent, Earl Goetheer, Ruud Kortlever
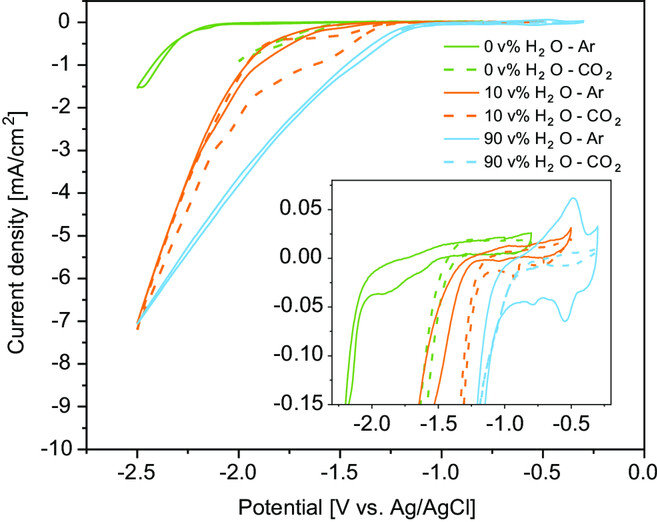
-
Vapor–liquid equilibrium (VLE) data for the binary systems tetrahydrofuran (THF) + acetic acid (AA) and THF + trichloroethylene (TCE) were measured under isobaric conditions using an ebulliometer. The boiling temperatures for the systems (THF + AA/THF + TCE) are reported for 13/15 compositions and five/six different pressures ranging from 50.2/60.0 to 101.1/101.3 kPa, respectively. The THF + AA system shows simple phase behavior with no azeotrope formation. The THF + TCE system does not exhibit azeotrope formation but seems to have a pinch point close to the pure end of TCE. The nonrandom two-liquid (NRTL) and universal quasichemical (UNIQUAC) activity coefficient models were used to accurately fit the binary (PTx) data. Both models were able to fit the binary VLE data satisfactorily. However, the NRTL model was found to be slightly better than UNIQUAC model in fitting the VLE data for both systems. The results can be used for designing liquid–liquid extraction and distillation processes involving mixtures of THF, AA, and TCE.
Vyomesh M. Parsana, Sachin Parikh, Keval Ziniya, Hirvita Dave, Piyush Gadhiya, Kedar Joshi, Dolly Gandhi, Thijs J. H. Vlugt, and Mahinder Ramdin
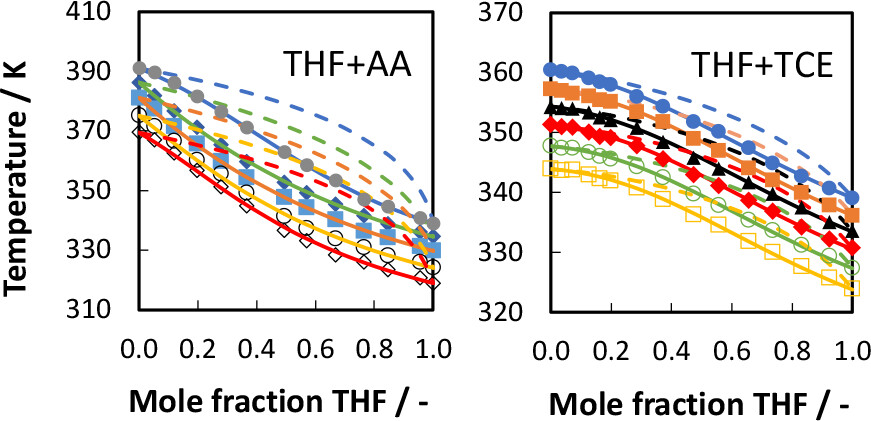
-
The use of gas diffusion electrodes that supply gaseous CO2 directly to the catalyst layer has greatly improved the performance of electrochemical CO2 conversion. However, reports of high current densities and Faradaic efficiencies primarily come from small lab scale electrolysers. Such electrolysers typically have a geometric area of 5 cm2, while an industrial electrolyser would require an area closer to 1 m2. The difference in scales means that many limitations that manifest only for larger electrolysers are not captured in lab scale setups. We develop a 2D computational model of both a lab scale and upscaled CO2 electrolyser to determine performance limitations at larger scales and how they compare to the performance limitations observed at the lab scale. We find that for the same current density larger electrolysers exhibit much greater reaction and local environment inhomogeneity. Increasing catalyst layer pH and widening concentration boundary layers of the KHCO3 buffer in the electrolyte channel lead to higher activation overpotential and increased parasitic loss of reactant CO2 to the electrolyte solution. We show that a variable catalyst loading along the direction of the flow channel may improve the economics of a large scale CO2 electrolyser.
J.W. Blake, V. Konderla, L.M. Baumgartner, D.A. Vermaas, J.T. Padding and J.W. Haverkort
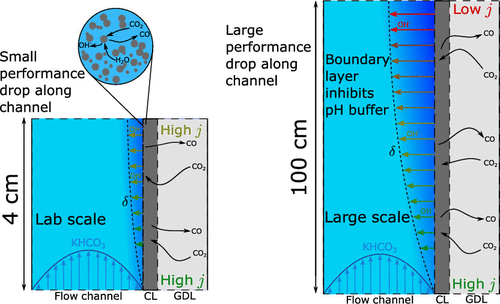
-
The electrochemical dinitrogen reduction reaction (NRR) has recently gained much interest as it can potentially produce ammonia from renewable intermittent electricity and replace the Haber–Bosch process. Previous literature studies report Fe- and Mo-carbides as promising electrocatalysts for the NRR with activities higher than other metals. However, recent understanding of extraneous ammonia and nitrogen oxide contaminations have challenged previously published results. Here, we critically assess the NRR performance of several Fe- and Mo-carbides reported as promising by implementing a strict experimental protocol to minimize the effect of impurities. The successful synthesis of α-Mo2C decorated carbon nanosheets, α-Mo2C nanoparticles, θ-Fe3C nanoparticles, and χ-Fe5C2 nanoparticles was confirmed by X-ray diffraction, scanning and transmission electron microscopy, and X-ray photoelectron and Mössbauer spectroscopy. After performing NRR chronoamperometric tests with the synthesized materials, the ammonia concentrations varied between 37 and 124 ppb and are in close proximity with the estimated ammonia background level. Notwithstanding the impracticality of these extremely low ammonia yields, the observed ammonia did not originate from the electrochemical nitrogen reduction but from unavoidable extraneous ammonia and NOx impurities. These findings are in contradiction with earlier literature studies and show that these carbide materials are not active for the NRR under the employed conditions. This further emphasizes the importance of a strict protocol in order to distinguish between a promising NRR catalyst and a false positive.
Boaz Izelaar, Davide Ripepi, Simone Asperti, A. Iulian Dugulan, Ruud W.A. Hendrikx, Amarante J. Böttger, Fokko M. Mulder and Ruud Kortlever
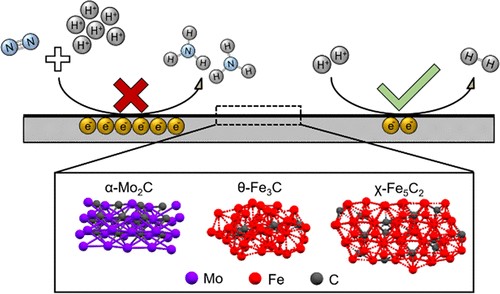
-
N-doped carbon materials can be efficient and cost-effective catalysts for the electrochemical CO2 reduction reaction (CO2RR). Activators are often used in the synthesis process to increase the specific surface area and porosity of these carbon materials. However, owing to the diversity of activators and the differences in physicochemical properties that these activators induce, the influence of activators used for the synthesis of N-doped carbon catalysts on their electrochemical performance is unclear. In this study, a series of bagasse-derived N-doped carbon catalysts is prepared with the assistance of different activators to understand the correlation between activators, physicochemical properties, and electrocatalytic performance for the CO2RR. The properties of N-doped carbon catalysts, such as N-doping content, microstructure, and degree of graphitization, are found to be highly dependent on the type of activator applied in the synthesis procedure. Moreover, the overall CO2RR performance of the synthesized electrocatalysts is not determined only by the N-doping level and the configuration of the N-dopant, but rather by the overall surface chemistry, where the porosity and the degree of graphitization are jointly responsible for significant differences in CO2RR performance.
Shilong Fu, Ming Li, Simone Asperti, Prof. Dr. Wiebren de Jong and Dr. Ruud Kortlever
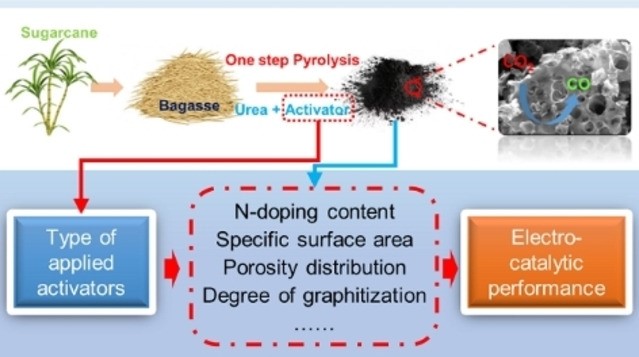
-
Microbial electrochemical technologies (METs) employ microorganisms utilizing solid-state electrodes as either electron sink or electron source, such as in microbial electrosynthesis (MES). METs reaction rate is traditionally normalized to the electrode dimensions or to the electrolyte volume, but should also be normalized to biomass amount present in the system at any given time. In biofilm-based systems, a major challenge is to determine the biomass amount in a non-destructive manner, especially in systems operated in continuous mode and using 3D electrodes. We developed a simple method using a nitrogen balance and optical density to determine the amount of microorganisms in biofilm and in suspension at any given time. For four MES reactors converting CO2 to carboxylates, >99% of the biomass was present as biofilm after 69 days of reactor operation. After a lag phase, the biomass-specific growth rate had increased to 0.12–0.16 days−1. After 100 days of operation, growth became insignificant. Biomass-specific production rates of carboxylates varied between 0.08–0.37 molC molX−1d−1. Using biomass-specific rates, one can more effectively assess the performance of MES, identify its limitations, and compare it to other fermentation technologies.
Marijn Winkelhorst, Oriol Cabau-Peinado, Adrie J.J. Straathof and Ludovic Jourdin
-
The electrochemical CO2 reduction reaction (CO2RR) is an attractive method to produce renewable fuel and chemical feedstock using clean energy sources. Formate production represents one of the most economical target products from CO2RR but is primarily produced using post-transition metal catalysts that require comparatively high overpotentials. Here a composition of bimetallic Cu–Pd is formulated on 2D Ti3C2Tx (MXene) nanosheets that are lyophilized into a highly porous 3D aerogel, resulting in formate production much more efficient than post-transition metals. Using a membrane electrode assembly (MEA), formate selectivities >90% are achieved with a current density of 150 mA cm−2 resulting in the highest ever reported overall energy efficiency of 47% (cell potentials of −2.8 V), over 5 h of operation. A comparable Cu-Pd aerogel achieves near-unity CO production without the MXene templating. This simple strategy represents an important step toward the experimental demonstration of 3D-MXenes-based electrocatalysts for CO2RR application and opens a new platform for the fabrication of macroscale aerogel MXene-based electrocatalysts.
M Abdinejad, S Subramanian, MK Motlagh, M Noroozifar, S Duangdangchote, I Neporozhnii, D Ripepi, D Pinto,aM Li, K Tang, J Middelkoop, A Urakawa, O Voznyy, HB Kraatz, T Burdyny.
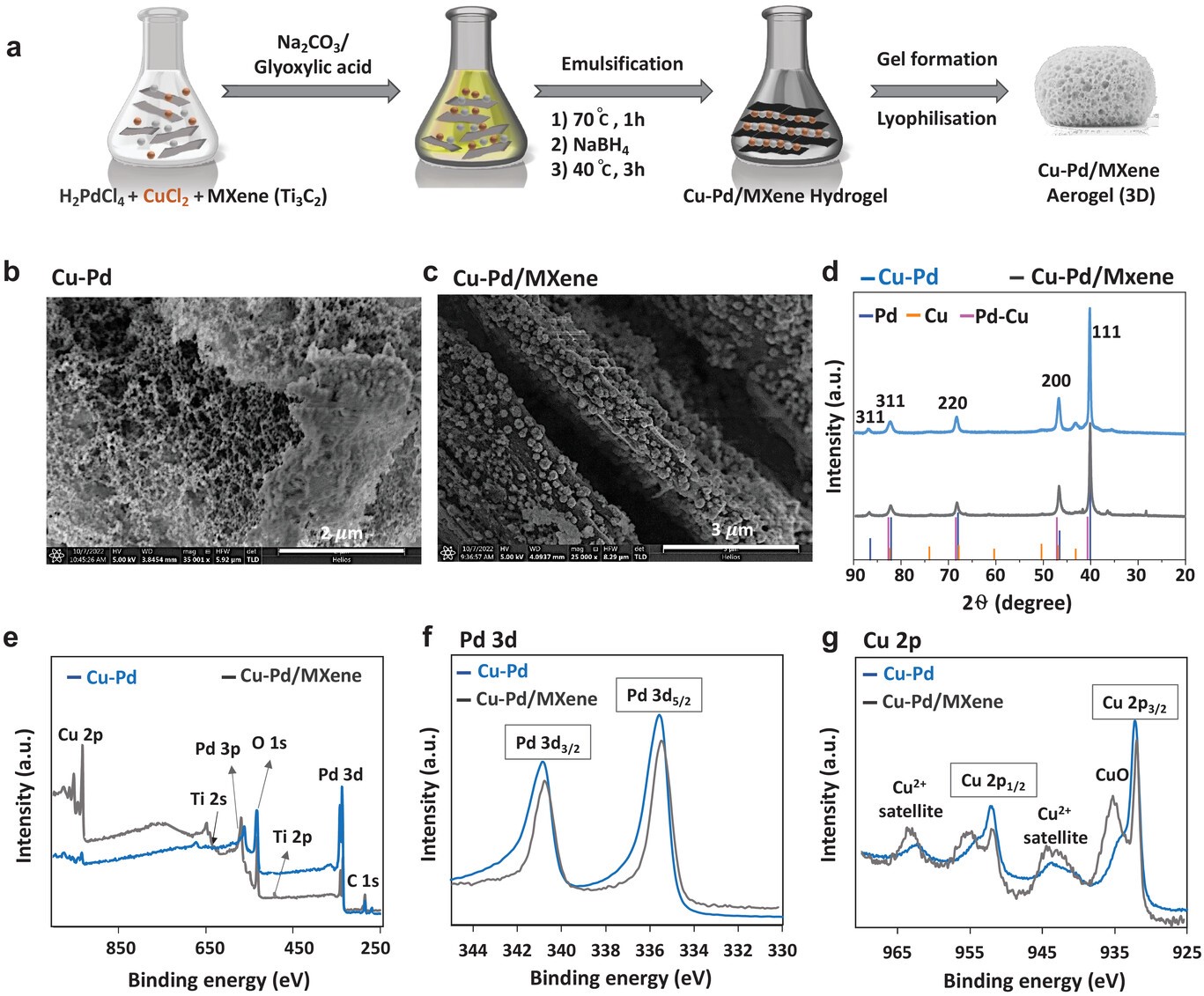
-
The electrochemical reduction of CO2 (CO2RR) on silver catalysts has been demonstrated under elevated current density, longer reaction times, and intermittent operation. Maintaining performance requires that CO2 can access the entire geometric catalyst area, thus maximizing catalyst utilization. Here we probe the time-dependent factors impacting geometric catalyst utilization for CO2RR in a zero-gap membrane electrode assembly. We use three flow fields (serpentine, parallel, and interdigitated) as tools to disambiguate cell behavior. Cathode pressure drop is found to play the most critical role in maintaining catalyst utilization at all time scales by encouraging in-plane CO2 transport throughout the gas-diffusion layer (GDL) and around salt and water blockages. The serpentine flow channel with the highest pressure drop is then the most failure-resistant, achieving a CO partial current density of 205 mA/cm2 at 2.76 V. These findings are confirmed through selectivity measurements over time, double-layer capacitance measurements to estimate GDL flooding, and transport modeling of the spatial CO2 concentration.
S Subramanian, K Yang, M Li, M Sassenburg, M Abdinejad, E Irtem, J Middelkoop, T Burdyny.
-
Salt precipitation is a problem in electrochemical CO2 reduction electrolyzers that limits their long-term durability and industrial applicability by reducing the active area, causing flooding and hindering gas transport. Salt crystals form when hydroxide generation from electrochemical reactions interacts homogeneously with CO2 to generate substantial quantities of carbonate. In the presence of sufficient electrolyte cations, the solubility limits of these species are reached, resulting in “salting out” conditions in cathode compartments. Detrimental salt precipitation is regularly observed in zero-gap membrane electrode assemblies, especially when operated at high current densities. This Perspective briefly discusses the mechanisms for salt formation, and recently reported strategies for preventing or reversing salt formation in zero-gap CO2 reduction membrane electrode assemblies. We link these approaches to the solubility limit of potassium carbonate within the electrolyzer and describe how each strategy separately manipulates water, potassium, and carbonate concentrations to prevent (or mitigate) salt formation.
M Sassenburg, M Kelly, S Subramanian, WA Smith, T Burdyny
-
The thermophysical properties of aqueous electrolyte solutions are of interest for applications such as water electrolyzers and fuel cells. Molecular dynamics (MD) and continuous fractional component Monte Carlo (CFCMC) simulations are used to calculate densities, transport properties (i.e., self-diffusivities and dynamic viscosities), and solubilities of H2 and O2 in aqueous sodium and potassium hydroxide (NaOH and KOH) solutions for a wide electrolyte concentration range (0–8 mol/kg). Simulations are carried out for a temperature and pressure range of 298–353 K and 1–100 bar, respectively. The TIP4P/2005 water model is used in combination with a newly parametrized OH– force field for NaOH and KOH. The computed dynamic viscosities at 298 K for NaOH and KOH solutions are within 5% from the reported experimental data up to an electrolyte concentration of 6 mol/kg. For most of the thermodynamic conditions (especially at high concentrations, pressures, and temperatures) experimental data are largely lacking. We present an extensive collection of new data and engineering equations for H2 and O2 self-diffusivities and solubilities in NaOH and KOH solutions, which can be used for process design and optimization of efficient alkaline electrolyzers and fuel cells.
Parsa Habibi, Ahmadreza Rahbari, Samuel Blazquez, Carlos Vega, Poulumi Dey, Thijs J. H. Vlugt, and Othonas A. Moultos
-
There is a growing interest in the development of routes to produce formic acid from CO2, such as the electrochemical reduction of CO2 to formic acid. The solubility of CO2 in the electrolyte influences the production rate of formic acid. Here, the dependence of the CO2 solubility in aqueous HCOOH solutions with electrolytes on the composition and the NaCl concentration was studied by Continuous Fractional Component Monte Carlo simulations at 298.15 K and 1 bar. The chemical potentials of CO2, H2O, and HCOOH were obtained directly from single simulations, enabling the calculation of Henry coefficients and subsequently considering salting in or salting out effects. As the force fields for HCOOH and H2O may not be compatible due to the presence of strong hydrogen bonds, the Gibbs–Duhem integration test was used to test this compatibility. The combination of the OPLS/AA force field with a new set of parameters, in combination with the SPC/E force field for water, was selected. It was found that the solubility of CO2 decreases with increasing NaCl concentration in the solution and increases with the increase of HCOOH concentration. This continues up to a certain concentration of HCOOH in the solution, after which the CO2 solubility is high and the NaCl concentration has no significant effect.
Dominika O. Wasik, H. Mert Polat, Mahinder Ramdin, Othonas A. Moultos, Sofia Calero, and Thijs J. H. Vlugt
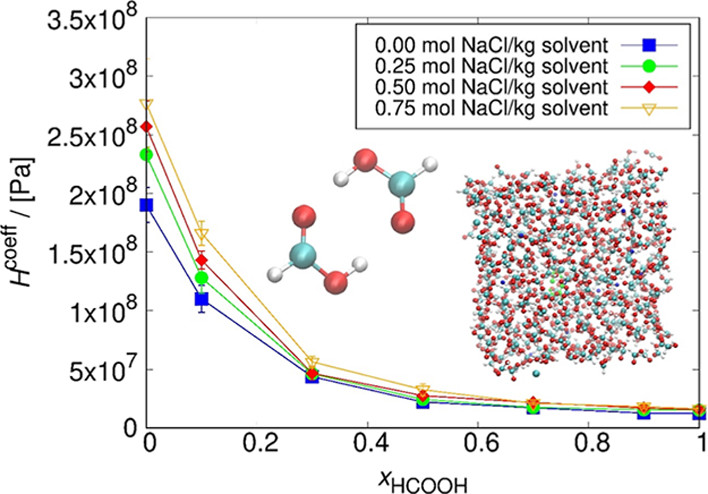
-
The electrochemical reduction of carbon dioxide (CO2) to value-added chemicals is a promising strategy to mitigate climate change. Metalloporphyrins have been used as a promising class of stable and tunable catalysts for the electrochemical reduction reaction of CO2 (CO2RR) but have been primarily restricted to single-carbon reduction products. Here, we utilize functionalized earth-abundant manganese tetraphenylporphyrin-based (Mn-TPP) molecular electrocatalysts that have been immobilized via electrografting onto a glassy carbon electrode (GCE) to convert CO2 with overall 94 % Faradaic efficiencies, with 62 % being converted to acetate. Tuning of Mn-TPP with electron-withdrawing sulfonate groups (Mn-TPPS) introduced mechanistic changes arising from the electrostatic interaction between the sulfonate groups and water molecules, resulting in better surface coverage, which facilitated higher conversion rates than the non-functionalized Mn-TPP. For Mn-TPP only carbon monoxide and formate were detected as CO2 reduction products. Density-functional theory (DFT) calculations confirm that the additional sulfonate groups could alter the C−C coupling pathway from *CO→*COH→*COH-CO to *CO→*CO-CO→*COH-CO, reducing the free energy barrier of C−C coupling in the case of Mn-TPPS. This opens a new approach to designing metalloporphyrin catalysts for two carbon products in CO2RR.
M. Abdinejad, T. Yuan, K. Tang, S. Duangdangchote, A. Farzi, H.P. Iglesias van Montfort, M. Li, J. Middelkoop, M. Wolff, A. Seifitokaldani, O. Voznyy, T. Burdyny
-
Electrochemical reduction of CO2 heavily depends on the reaction conditions found near the electrode surface. These local conditions are affected by phenomena such as electric double layer formation and steric effects of the solution species, which in turn impact the passage of CO2 molecules to the catalytic surface. Most models for CO2 reduction ignore these effects, leading to an incomplete understanding of the local electrode environment. In this work, we present a modeling approach consisting of a set of size-modified Poisson–Nernst–Planck equations and the Frumkin interpretation of Tafel kinetics. We introduce a modification to the steric effects inside the transport equations which results in more realistic concentration profiles. We also show how the modification lends the model numerical stability without adopting any separate stabilization technique. The model can replicate experimental current densities and faradaic efficiencies till −1.5 vs. SHE/V of applied electrode potential. We also show the utility of this approach for systems operating at elevated CO2 pressures. Using Frumkin-corrected kinetics gels well with the theoretical understanding of the double layer. Hence, this work provides a sound mechanistic understanding of the CO2 reduction process, from which new insights on key performance controlling parameters can be obtained.
Esaar Naeem Butt, Johan T. Padding and Remco Hartkamp
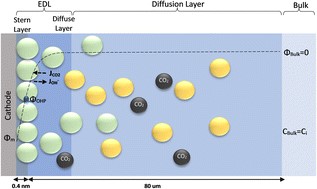
-
Membraneless parallel-plate electrolyzers use electrolyte flow to avoid product crossover. Using a mixture model neglecting inertia, and assuming an exponential gas fraction profile, we derive approximate analytical expressions for the velocity profile and pressure drop for thin plumes. We verify these expressions using numerical solutions obtained with COMSOL and validate them using experimental data from the literature. We find that the wall gas fraction increases rapidly at small heights, but becomes fairly constant at larger heights. These expressions serve as a guiding framework for designing a membraneless parallel-plate electrolyzer by quantifying the maximum possible height. We find that buoyancy driven membraneless parallel-plate electrolyzers with a 3 mm gap can be designed with a maximum height of around 7.6 cm at 1000 A/m2 for operation with 98% product purity at atmospheric pressure. For a forced flow at Re=1000, the same electrolyzer can be made around 17.6 cm tall at 1000 A/m2. These limits can be further improved with smaller bubbles or higher pressure.
A. Rajora and J.W. Haverkort
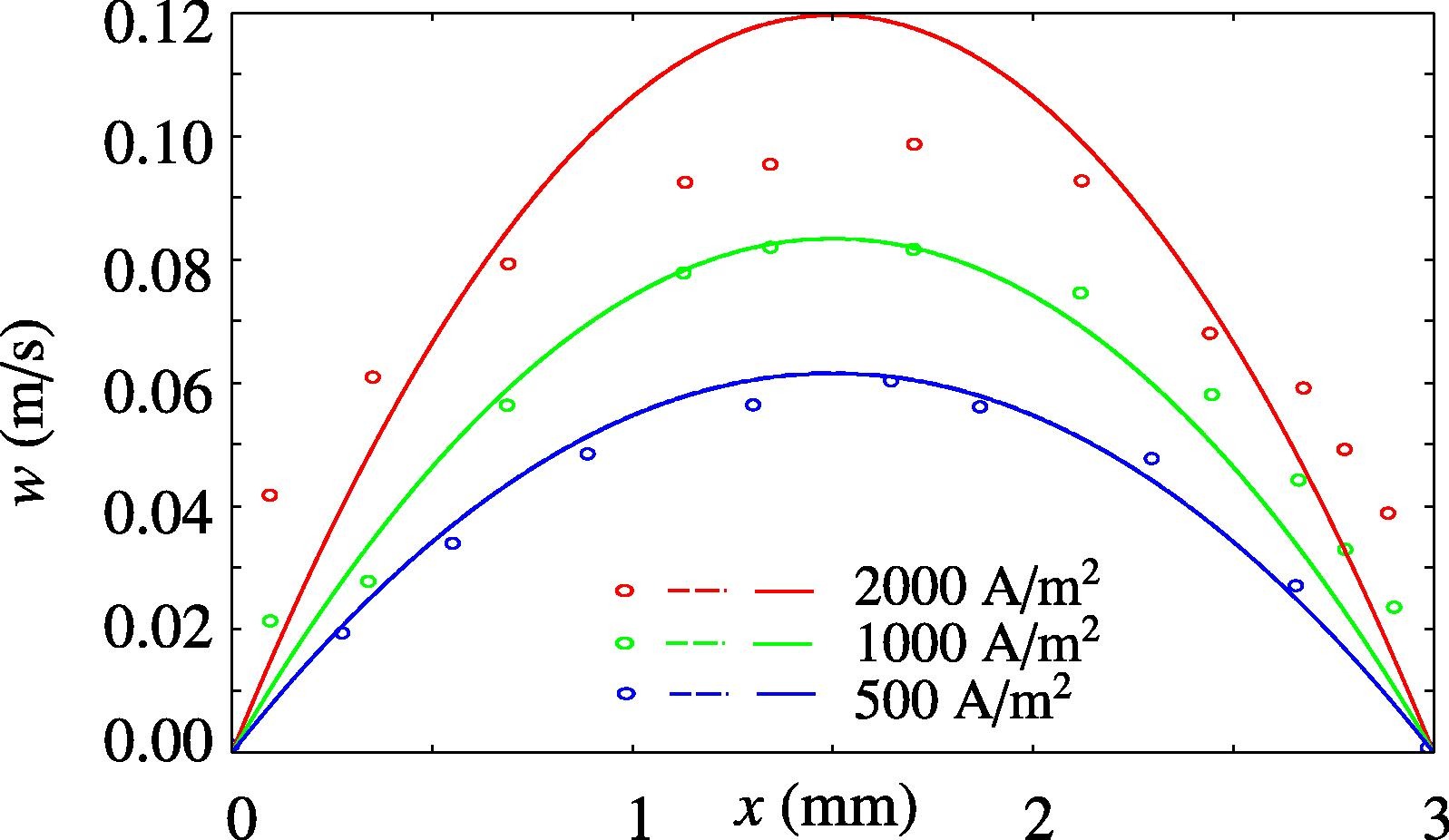
-
Integrating carbon dioxide (CO2) electrolysis with CO2 capture provides exciting new opportunities for energy reductions by simultaneously removing the energy-demanding regeneration step in CO2 capture and avoiding critical issues faced by CO2 gas-fed electrolysers. However, understanding the potential energy advantages of an integrated process is not straightforward due to the interconnected processes which require knowledge of both capture and electrochemical conversion processes. Here, we identify the upper limits of the integrated process from an energy perspective by comparing the working principles and performance of integrated and sequential approaches. Our high-level energy analyses unveil that an integrated electrolyser must show similar performance to the gas-fed electrolyser to ensure an energy benefit of up to 44% versus the sequential route. However, such energy benefits diminish if future gas-fed electrolysers resolve the CO2 utilisation issue and if an integrated electrolyser shows lower conversion efficiencies than the gas-fed system.
Mengran Li, Erdem Irtem, Hugo-Pieter Iglesias van Montfort, Maryam Abdinejad & Thomas Burdyny
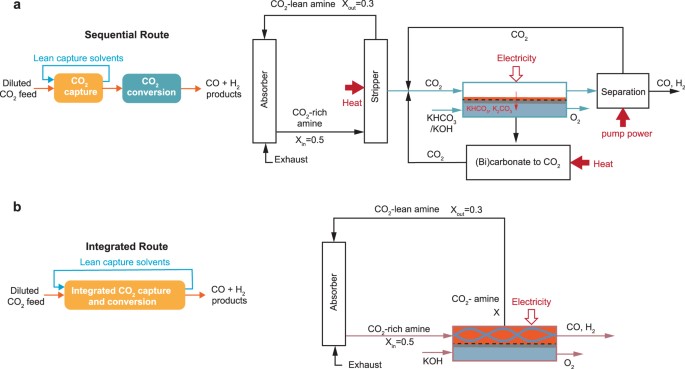
-
Electrochemical reduction of CO2 using renewable energy is a promising avenue for sustainable production of bulk chemicals. However, CO2 electrolysis in aqueous systems is severely limited by mass transfer, leading to low reactor performance insufficient for industrial application. This paper shows that structured reactors operated under gas–liquid Taylor flow can overcome these limitations and significantly improve the reactor performance. This is achieved by reducing the boundary layer for mass transfer to the thin liquid film between the CO2 bubbles and the electrode. This work aims to understand the relationship between process conditions, mass transfer, and reactor performance by developing an easy-to-use analytical model. We find that the film thickness and the volume ratio of CO2/electrolyte fed to the reactor significantly affect the current density and the faradaic efficiency. Additionally, we find industrially relevant performance when operating the reactor at an elevated pressure beyond 5 bar. We compare our predictions with numerical simulations based on the unit cell approach, showing good agreement for a large window of operating parameters, illustrating when the easy-to-use predictive expressions for the current density and faradaic efficiency can be applied.
Isabell Bagemihl, Chaitanya Bhatraju, J. Ruud van Ommen and Volkert van Steijn
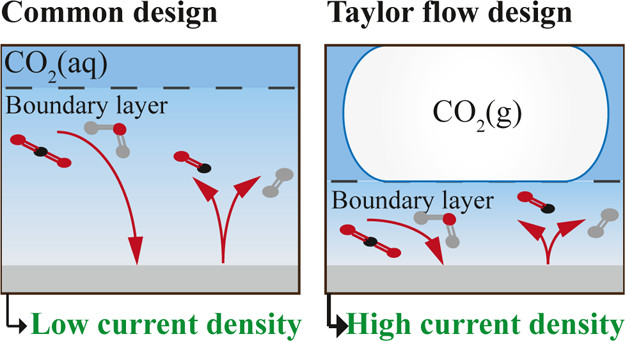
-
Mass transport of different species plays a crucial role in electrochemical conversion of CO2 due to the solubility limit of CO2 in aqueous electrolytes. In this study, we investigate the transport of CO2 and other ionic species through the electrolyte and the membrane, and its impact on the scale-up process of HCOO−/HCOOH formation. The mass transport of ions to the electrode and the membrane is modelled at constant current density. The mass transport limitations of CO2 on the formation of HCOO−/HCOOH is investigated at different pressures ranges from 5–40 bar. The maximum achievable partial current density of formate/formic acid is increased with increasing CO2 pressure. We use an ion exchange membrane model to understand the ion transport behaviour for both the monopolar and bipolar membranes. The cation exchange (CEM) and anion exchange membrane (AEM) model show that ion transport is limited by the electrolyte salt concentrations. For 0.1 M KHCO3, the AEM reaches the limiting current density more quickly than the CEM. For the BPM model, ion transport across the diffusion layer on either side of the BPM is also included to understand the concentration polarization across the BPM. The model revealed that the polarization losses across the bipolar membrane depend on the pH of the electrolyte used for the CO2 reduction reaction (CO2RR). The polarization loss on the anolyte side decreases with an increasing pH, while, on the cathode side, it increases with increasing catholyte pH. With this combined model for the electrode reactions and the membrane transport, we are able to account for the various factors influencing the polarization losses in the CO2 electrolyzer. To complete the analysis, we simulated the full cell polarization curve and fitted with the experimental data.
Selvaraj Chinnathambi, Mahinder Ramdin and Thijs J. H. Vlugt
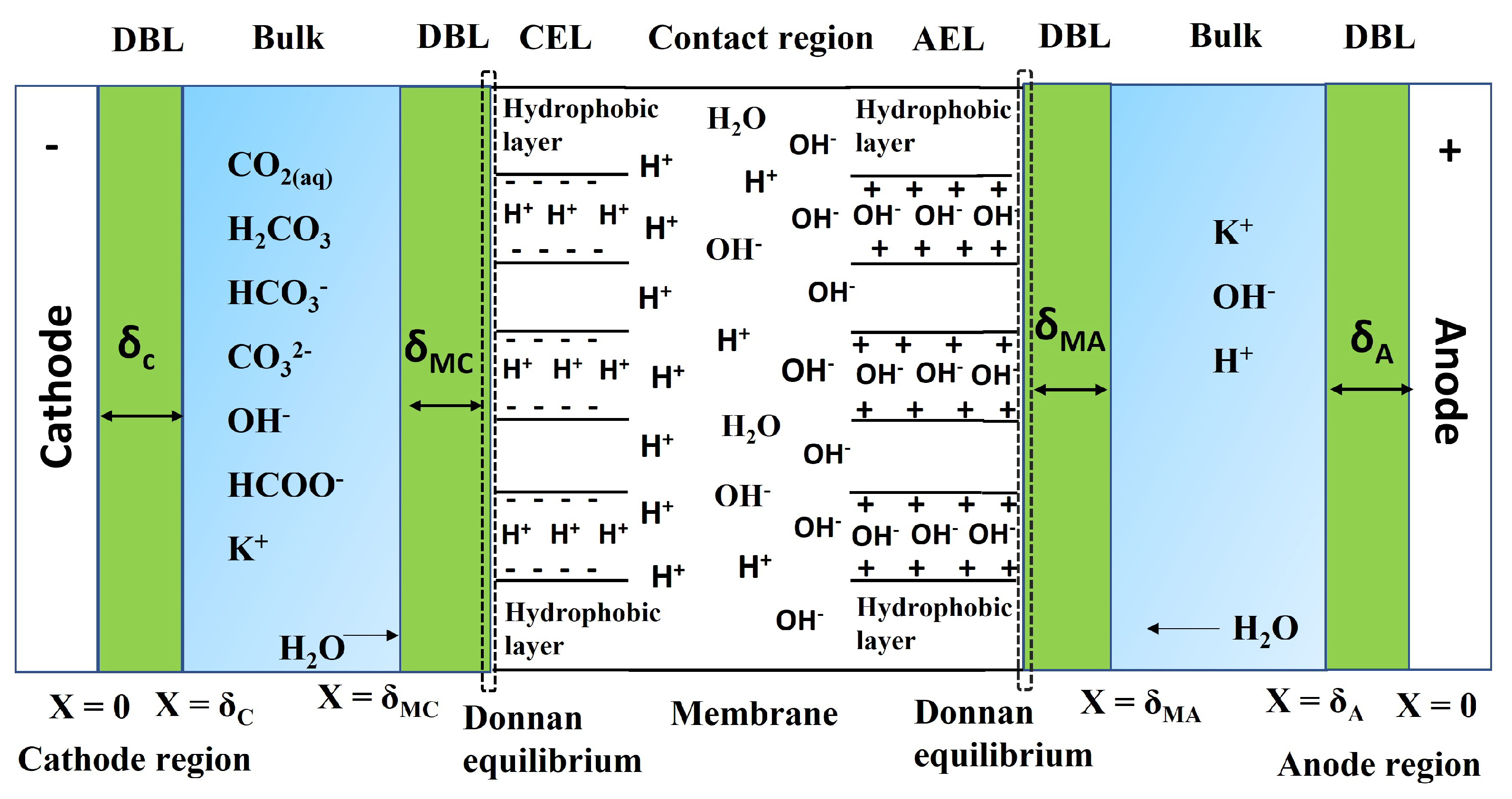
-
One of the important parameters in water management of proton exchange membranes is the electro-osmotic drag (EOD) coefficient of water. The value of the EOD coefficient is difficult to justify, and available literature data on this for Nafion membranes show scattering from in experiments and simulations. Here, we use a classical all-atom model to compute the EOD coefficient and thermodynamic properties of water from molecular dynamics simulations for temperatures between 330 and 420 K, and for different water contents between λ = 5 and λ = 20. λ is the ratio between the moles of water molecules to the moles of sulfonic acid sites. This classical model does not capture the Grotthuss mechanism; however, it is shown that it can predict the EOD coefficient within the range of experimental values for λ = 5 where the vehicular mechanism dominates proton transfer. For λ > 5, the Grotthuss mechanism becomes dominant. To obtain the EOD coefficient, average velocities of water and ions are computed by imposing different electric fields to the system. Our results show that the velocities of water and hydronium scale linearly with the electric field, resulting in a constant ratio of ca. 0.4 within the error bars. We find that the EOD coefficient of water linearly increases from 2 at λ = 5 to 8 at λ = 20 and the results are not sensitive to temperature. The EOD coefficient at λ = 5 is within the range of experimental values, confirming that the model can capture the vehicular transport of protons well. At λ = 20, due to the absence of proton hopping in the model, the EOD coefficient is overestimated by a factor of 3 compared to experimental values. To analyze the interactions between water and Nafion, the partial molar enthalpies and partial molar volumes of water are computed from molecular dynamics simulations. At different water uptakes, multiple linear regression is used on raw simulation data within a narrow composition range of water inside the Nafion membrane. The partial molar volumes and partial molar excess enthalpies of water asymptotically approach the molar volumes and molar excess enthalpies of pure water for water uptakes above 5. This confirms the model can capture the bulklike behavior of water in the Nafion at high water uptakes.
Ahmadreza Rahbari, Remco Hartkamp, Othonas A. Moultos, Albert Bos, Leo J. P. van den Broeke, Mahinder Ramdin, David Dubbeldam, Alexey V. Lyulin, and Thijs J. H. Vlugt
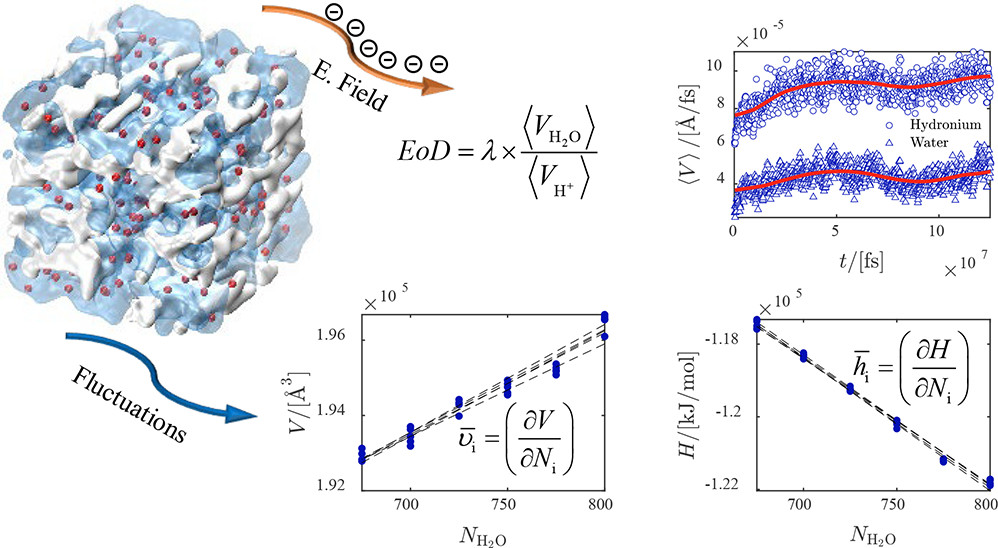
-
The selective separation of ions is a major technological challenge having far-ranging impacts from product separation in electrochemical production of base chemicals from CO2 to water purification. In recent years, ion-selective electrochemical systems leveraging redox-materials emerged as an attractive platform based on their reversibility and remarkable ion selectivity. In the present study, we present an ultrasound-intensified fabrication process for polyvinyl ferrocene (PVF)–functionalized electrodes in a carbon nanotube (CNT) matrix for selective electro-adsorption of formate ions. To this end, a response surface methodology involving the Box–Behnken design with three effective independent variables, namely, PVF to CNT ratio, sonication duration, and ultrasonic amplitude was applied to reach the maximum formate adsorption efficiency. The fabricated electrodes were characterized using cyclic voltammetry.
Sevgi Polat, Ruud Kortlever, Huseyin Burak Eral
-
The electrochemical CO2 reduction reaction (CO2RR) is important for a sustainable future. Key insights into the reaction pathways have been obtained by density functional theory (DFT) analysis, but so far, DFT has been unable to give an overall understanding of selectivity trends without important caveats. We show that an unconsidered parameter in DFT models of electrocatalysts─the surface coverage of reacting species─is crucial for understanding the CO2RR selectivities for different surfaces. Surface coverage is a parameter that must be assumed in most DFT studies of CO2RR electrocatalysts, but so far, only the coverage of nonreacting adsorbates has been treated. Explicitly treating the surface coverage of reacting adsorbates allows for an investigation that can more closely mimic operating conditions. Furthermore, and of more immediate importance, the use of surface coverage-dependent adsorption energies allows for the extraction of ratios of adsorption energies of CO2RR intermediates (COOHads and HCOOads) that are shown to be predictive of selectivity and are not susceptible to systematic errors. This approach allows for categorization of the selectivity of several monometallic catalysts (Pt, Pd, Au, Ag, Zn, Cu, Rh, W, Pb, Sn, In, Cd, and Tl), even problematic ones such as Ag or Zn, and does so by only considering the adsorption energies of known intermediates. The selectivity of the further reduction of COOHads can now be explained by a preference for Tafel or Heyrovsky reactions, recontextualizing the nature of selectivity of some catalysts. In summary, this work resolves differences between DFT and experimental studies of the CO2RR and underlines the importance of surface coverage.
Andrew R. T. Morrison, Mahinder Ramdin, Leo J. P. van der Broeke, Wiebren de Jong, Thijs J. H. Vlugt, and Ruud Kortlever

-
Copper is one of the most promising catalysts for the CO2 reduction reaction (CO2RR) due to its unique capability of producing multicarbon products in appreciable quantities. Most of the CO2RR research efforts have been directed towards the development of new electrocatalysts to either increase product selectivities or decrease overpotentials. In contrast, only a few studies have systematically tested or benchmarked CO2RR performances of electrocatalysts. In this paper, for the first time, the performances of five different polycrystalline copper foils purchased from different suppliers are benchmarked for their CO2RR performance. Their differences are characterized in terms of microstructural features and the effect that these microstructural properties have on the electrocatalytic behavior during potentiostatic CO2RR experiments are evaluated. It is shown that the potential applied is the dominant factor controlling CO2RR selectivities, leading to the conclusion that microstructural properties of polycrystalline copper electrodes have a negligible effect on the outcome of CO2RR experiments.
Simone Asperti, Dr. Ruud Hendrikx, Dr. Yaiza Gonzalez-Garcia, Dr. Ruud Kortlever

-
Electrolysis of water, CO2, and nitrogen-based compounds presents the opportunity of generating fossil-free fuels and feedstocks at an industrial scale. Such devices are complex in operation, and their performance metrics are usually reported as electrode-averaged quantities. In this work, we report the usage of infrared thermography to map the electrochemical activity of a gas-diffusion electrode performing water and CO2 reduction. By associating the heat map to a characteristic catalytic activity, the presented system can capture electrochemical and physical phenomena as they occur in electrolyzers for large-scale energy applications. We demonstrate applications for catalyst screening, catalyst-degradation measurements, and spatial activity mapping for water and CO2 electrolysis at current densities up to 0.2 A cm–2. At these current densities we report catalyst temperature increases (>10 K for 0.2 A cm–2) not apparent otherwise. Furthermore, substantial localized current density fluctuations are present. These observations challenge assumed local conditions, providing new fundamental and applied perspectives.
Hugo-Pieter Iglesias van Montfort and Thomas Burdyny

-
The electrochemical reduction of carbon dioxide (CO2) to value-added materials has received considerable attention. Both bulk transition-metal catalysts and molecular catalysts affixed to conductive noncatalytic solid supports represent a promising approach toward the electroreduction of CO2. Here, we report a combined silver (Ag) and pyridine catalyst through a one-pot and irreversible electrografting process, which demonstrates the enhanced CO2 conversion versus individual counterparts. We find that by tailoring the pyridine carbon chain length, a 200 mV shift in the onset potential is obtainable compared to the bare silver electrode. A 10-fold activity enhancement at −0.7 V vs reversible hydrogen electrode (RHE) is then observed with demonstratable higher partial current densities for CO, indicating that a cocatalytic effect is attainable through the integration of the two different catalytic structures. We extended the performance to a flow cell operating at 150 mA/cm2, demonstrating the approach’s potential for substantial adaptation with various transition metals as supports and electrografted molecular cocatalysts.
Maryam Abdinejad, Erdem Irtem, Amirhossein Farzi, Mark Sassenburg, Siddhartha Subramanian, Hugo-Pieter Iglesias van Montfort, Davide Ripepi, Mengran Li, Joost Middelkoop, Ali Seifitokaldani and Thomas Burdyny
-
Continued advancements in the electrochemical reduction of CO2 (CO2RR) have emphasized that reactivity, selectivity, and stability are not explicit material properties but combined effects of the catalyst, double-layer, reaction environment, and system configuration. These realizations have steadily built upon the foundational work performed for a broad array of transition metals performed at 5 mA cm–2, which historically guided the research field. To encompass the changing advancements and mindset within the research field, an updated baseline at elevated current densities could then be of value. Here we seek to re-characterize the activity, selectivity, and stability of the five most utilized transition metal catalysts for CO2RR (Ag, Au, Pd, Sn, and Cu) at elevated reaction rates through electrochemical operation, physical characterization, and varied operating parameters to provide a renewed resource and point of comparison. As a basis, we have employed a common cell architecture, highly controlled catalyst layer morphologies and thicknesses, and fixed current densities. Through a dataset of 88 separate experiments, we provide comparisons between CO-producing catalysts (Ag, Au, and Pd), highlighting CO-limiting current densities on Au and Pd at 72 and 50 mA cm–2, respectively. We further show the instability of Sn in highly alkaline environments, and the convergence of product selectivity at elevated current densities for a Cu catalyst in neutral and alkaline media. Lastly, we reflect upon the use and limits of reaction rates as a baseline metric by comparing catalytic selectivity at 10 versus 200 mA cm–2. We hope the collective work provides a resource for researchers setting up CO2RR experiments for the first time.
Mark Sassenburg, Reinier de Rooij, Nathan T. Nesbitt, Recep Kas, Sanjana Chandrashekar, Nienke J. Firet, Kailun Yang, Kai Liu, Marijn A. Blommaert, Martin Kolen, Davide Ripepi, Wilson A. Smith and Thomas Burdyny
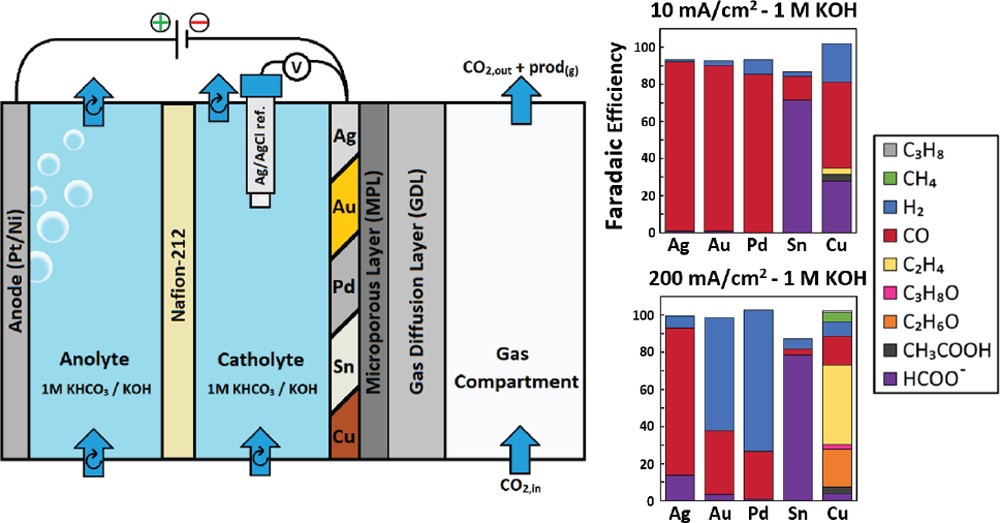
-
Incorporating (operational) flexibility into process design has been a key approach to cope with uncertainties. The increasing penetration of renewables and the need for developing new low-carbon technologies will increase the demand for flexibility in chemical processes. This paper presents a state-of-the-art review focusing on the origin, definition, and elements of flexibility in the chemical engineering context. The article points out a significant overlap in terminology and concepts, making it difficult to understand and compare flexibility potential and constraints among studies. Further, the paper identifies a lack of available metrics for assessing specific types of flexibility and the need for developing indicators for exploring the potential flexibility of novel chemical processes. The paper proposes a classification of flexibility types and provides an overview of design strategies that have been adopted so far to enable different types of flexibility. Finally, it offers a conceptual framework that can support designers to evaluate specific types of flexibility in early-stage assessments of novel chemical processes.
Martin Kolen, Davide Ripepi, Wilson A. Smith, Thomas Burdyny and Fokko M. Mulder
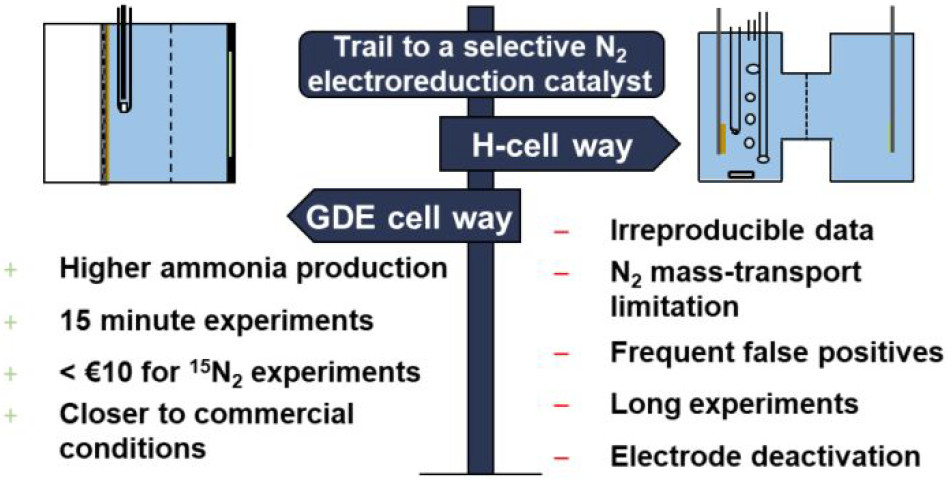
-
The ever-growing level of carbon dioxide (CO2) in our atmosphere, is at once a threat and an opportunity. The development of sustainable and cost-effective pathways to convert CO2 to value-added chemicals is central to reducing its atmospheric presence. Electrochemical CO2 reduction reactions (CO2RRs) driven by renewable electricity are among the most promising techniques to utilize this abundant resource; however, in order to reach a system viable for industrial implementation, continued improvements to the design of electrocatalysts is essential to improve the economic prospects of the technology. This review summarizes recent developments in heterogeneous porphyrin-based electrocatalysts for CO2 capture and conversion. We specifically discuss the various chemical modifications necessary for different immobilization strategies, and how these choices influence catalytic properties. Although a variety of molecular catalysts have been proposed for CO2RRs, the stability and tunability of porphyrin-based catalysts make their use particularly promising in this field. We discuss the current challenges facing CO2RRs using these catalysts and our own solutions that have been pursued to address these hurdles.
Maryam Abdinejad, Keith Tang, Caitlin Dao, Saeed Saedy and Tom Burdyny
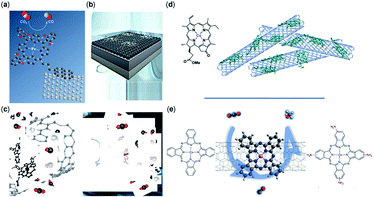
-
Carbon-based products are crucial to our society, but their production from fossil-based carbon is unsustainable. Production pathways based on the reuse of CO2 will achieve ultimate sustainability. Furthermore, the costs of renewable electricity production are decreasing at such a high rate, that electricity is expected to be the main energy carrier from 2040 onward. Electricity-driven novel processes that convert CO2 into chemicals need to be further developed. Microbial electrosynthesis is a biocathode-driven process in which electroactive microorganisms derive electrons from solid-state electrodes to catalyze the reduction of CO2 or organics and generate valuable extracellular multicarbon reduced products. Microorganisms can be tuned to high-rate and selective product formation. Optimization and upscaling of microbial electrosynthesis to practical, real life applications is dependent upon performance improvement while maintaining low cost. Extensive biofilm development, enhanced electron transfer rate from solid-state electrodes to microorganisms and increased chemical production rate require optimized microbial consortia, efficient reactor designs, and improved cathode materials.
Victoria Flexer and Ludovic Jourdin
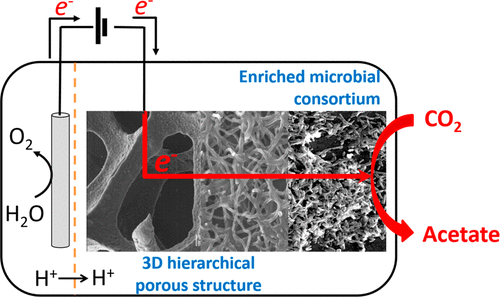
-
Up to now, computational modeling of microbial electrosynthesis (MES) has been underexplored, but is necessary to achieve breakthrough understanding of the process-limiting steps. Here, a general framework for modeling microbial kinetics in a MES reactor is presented. A thermodynamic approach is used to link microbial metabolism to the electrochemical reduction of an intracellular mediator, allowing to predict cellular growth and current consumption. The model accounts for CO2 reduction to acetate, and further elongation to n-butyrate and n-caproate. Simulation results were compared with experimental data obtained from different sources and proved the model is able to successfully describe microbial kinetics (growth, chain elongation, and product inhibition) and reactor performance (current density, organics titer). The capacity of the model to simulate different system configurations is also shown. Model results suggest CO2 dissolved concentration might be limiting existing MES systems, and highlight the importance of the delivery method utilized to supply it. Simulation results also indicate that for biofilm-driven reactors, continuous mode significantly enhances microbial growth and might allow denser biofilms to be formed and higher current densities to be achieved.
Oriol Cabau-Peinado, Adrie J. J. Straathof and Ludovic Jourdin
-
In the past decade, research in the field of microbial electrosynthesis (MES) has been driven forward by the development of cathode materials, electroactive bacteria or microbiome enrichment, and productivity improvements.
As the close of three complete funding cycles for the field is reached, recent reviews have sought to refocus emphasis to the eventual application of MES; a means of measurably reducing CO2 waste via the formation of valuable products.
Using present knowledge of bioelectrochemistry, and by learning lessons from adjacent fields, it becomes apparent that the simplest gains in performance are likely to come from advancements in the reactor rather than the biocatalysts. Varying the reactor and operating conditions of the system, however, require adapting these biocatalysts.
Ludovic Jourdin and Thomas Burdyny
-
The effect of different alkali metal cations on the oxygen evolution activity and battery capacity of nickel electrodes has recently been studied in low concentration alkali hydroxide electrolytes. As high concentration hydroxide electrolytes are favored in applications due to their high conductivity, we investigate if the cation effects observed in low concentration electrolytes translate to more industrially relevant conditions for both alkaline water electrolysis and nickel iron batteries. We investigate the alkali metal cation effect on the electrochemical properties of nickel electrodes in highly concentrated hydroxide electrolytes by adding Li+, Na+, Cs+ and Rb+ cations to a 6.5 M KOH electrolyte, while keeping the hydroxide concentration constant. For OER we find a trend in activity similar to that at low concentrations Rb+>Cs+>K+>Na+>Li+, where especially larger additions of Rb+ and Cs+ (1 M or 0.5 M) cause a significant decrease in OER potential. Smaller cations interact with the layered hydroxide structures in NiOOH to stabilize the α/γ phases and increase the potential for OER. The intercalation of small cations also causes an increase in battery electrode capacity because of the higher average valence of the Ni(OH)2/NiOOH α/γ pair. Small concentrations of Li+ added to a concentrated KOH electrolyte can therefore be beneficial for the nickel electrode battery functionality and for an integrated battery and electrolyser system, where it increases the battery capacity without a significant increase in OER onset potential.
A.Mangel Raventos, R.Kortlever
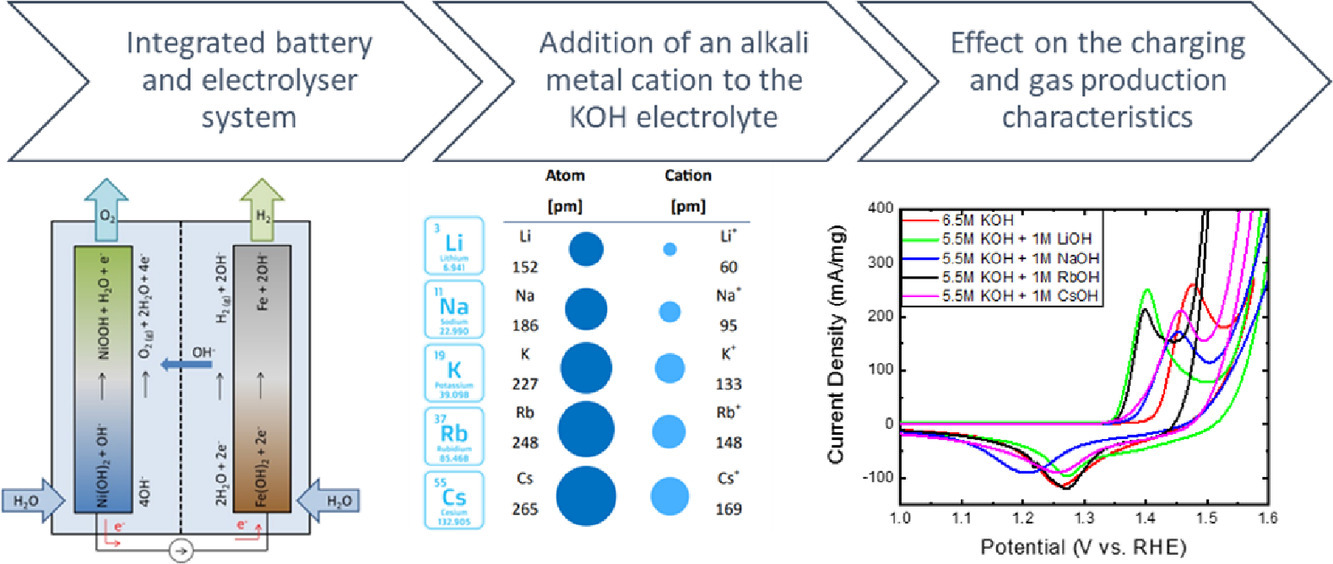
-
Rapid advances in electrocatalytic ammonia synthesis are impeded by laborious detection methods commonly used in the field and by constant risk of external contaminations, which generates misleading false positives. We developed a facile real-time GC-MS method for sensitive isotope NH3 quantification, requiring no external sample manipulations. This method ensures high detection reliability paramount to accelerate (electro-)catalyst screening.
Davide Ripepi, Riccardo Zaffaroni, Martin Kolen, Joost Middelkoop and Fokko M. Mulder
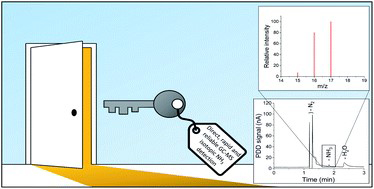
-
Finding alternative ways to tailor the electronic properties of a catalyst to actively and selectively drive reactions of interest has been a growing research topic in the field of electrochemistry. In this Letter, we investigate the tuning of the surface electronic properties of electrocatalysts via polymer modification. We show that when a nickel oxide water oxidation catalyst is coated with polytetrafluoroethylene, stable Ni–CFx bonds are introduced at the nickel oxide/polymer interface, resulting in shifting of the reaction selectivity away from the oxygen evolution reaction and toward hydrogen peroxide formation. It is shown that the electron-withdrawing character of the surface fluorocarbon molecule leaves a slight positive charge on the water oxidation intermediates at the adjacent active nickel sites, making their bonds weaker. The concept of modifying the surface electronic properties of an electrocatalyst via stable polymer modification offers an additional route to tune multipathway reactions in polymer/electrocatalyst environments, like with ionomer-modified catalysts or with membrane electrode assemblies.
Anirudh Venugopal, Laurentius H. T. Egberts, Jittima Meeprasert, Evgeny A. Pidko, Bernard Dam, Thomas Burdyny, Vivek Sinha, Wilson A. Smith
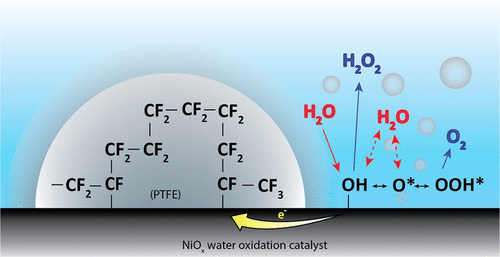
-
The specific identity of electrolyte cations has many implications in various electrochemical reactions. However, the exact mechanism by which cations affect electrochemical reactions is not agreed upon in the literature. In this report, we investigate the role of cations during the electrochemical reduction of CO2 by chelating the cations with cryptands, to change the interaction of the cations with the components of the electric double layer. As previously reported we do see the apparent suppression of CO2 reduction in the absence of cations. However, using in situ-SEIRAS we see that CO2 reduction does indeed take place albeit at very reduced scales. We also observe that cations play a role in tuning the absorption strengths of not only CO2 as has been speculated, but also that of reaction products such as CO.
S Chandrashekar, HP van Montfort, D Bohra, G Filonenko, H Geerlings, T Burdyny, WA Smith

-
The electrochemical reduction of bicarbonate to renewable chemicals without external gaseous CO2 supply has been motivated as a means of integrating conversion with upstream CO2 capture. The way that CO2 is formed and transported during CO2-mediated bicarbonate reduction in flow cells is profoundly different from conventional CO2 saturated and gas-fed systems and a thorough understanding of the process would allow further advancements. Here, we report a comprehensive two-phase mass transport model to estimate the local concentration of species in the porous electrode resultant from homogeneous and electrochemical reactions of (bi)carbonate and CO2. The model indicates that significant CO2 is generated in the porous electrode during electrochemical reduction, even though the starting bicarbonate solution contains negligible CO2. However, the in situ formation of CO2 and subsequent reduction to CO exhibits a plateau at high potentials due to neutralization of the protons by the alkaline reaction products, acting as the limiting step toward higher CO current densities. Nevertheless, the pH in the catalyst layer exhibits a relatively smaller rise, compared to conventional electrochemical CO2 reduction cells, because of the reaction between protons and CO32– and OH– that is confined to a relatively small volume. A large fraction of the CL exhibits a mildly alkaline environment at high current densities, while an appreciable amount of carbonic acid (0.1–1 mM) and a lower pH exist adjacent to the membrane, which locally favor hydrogen evolution, especially at low electrolyte concentrations. The results presented here provide insights into local cathodic conditions for both bicarbonate cells and direct-CO2 reduction membrane electrode assembly cells utilizing cation exchange membranes facing the cathode.
Kas R, Yang K, Yewale G, Crow A, Burdyny T, Smith WA
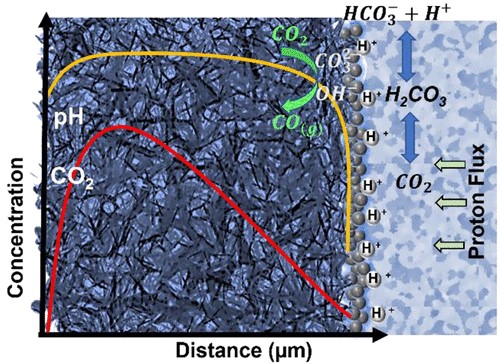
-
CO2 electrochemical reduction reaction (CO2RR) enables conversion of greenhouse gas CO2 into value-added products, which simultaneously reduces carbon emissions and reduces the usage of fossil fuels as feed materials to produce fuel/chemical products. It also provides the potential to integrate electrocatalytic processes with electricity from renewable sources for storing renewable energy. Membrane-electrode assemblies (MEAs) can be an efficient solution to address the key issues in aqueous electrolyzer design and enable the industrial scale-up. In this paper, we reviewed recent advances in the experimental design and simulation of MEAs. The discussion of existing challenges and future research priorities for guiding MEA development and understanding reaction mechanism is also provided.
Ge L, Rabiee H, Li M, Subramanian S, Zheng Y, Lee JH, Burdyny T, Wang H.
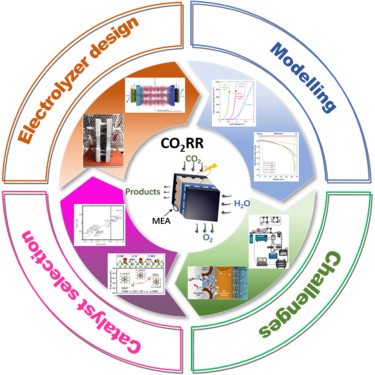
-
Surface conductivity in the electrical double layer (EDL) is known to be affected by proton hopping and diffusion at solid-liquid interfaces. Yet, the role of surface protolysis and its kinetics on the thermodynamic and transport properties of the EDL are usually ignored as physical models consider static surfaces. Here, using a novel molecular dynamics method mimicking surface protolysis, we unveil the impact of such chemical events on the system’s response. Protolysis is found to strongly affect the EDL and electrokinetic aspects with major changes in ζ potential and electro-osmotic flow.
Max F. Döpke, Fenna Westerbaan van der Meij, Benoit Coasne, and Remco Hartkamp
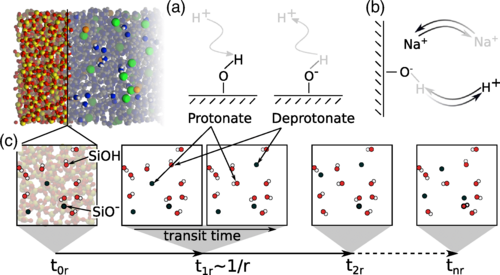
-
Incorporating (operational) flexibility into process design has been a key approach to cope with uncertainties. The increasing penetration of renewables and the need for developing new low-carbon technologies will increase the demand for flexibility in chemical processes. This paper presents a state-of-the-art review focusing on the origin, definition, and elements of flexibility in the chemical engineering context. The article points out a significant overlap in terminology and concepts, making it difficult to understand and compare flexibility potential and constraints among studies. Further, the paper identifies a lack of available metrics for assessing specific types of flexibility and the need for developing indicators for exploring the potential flexibility of novel chemical processes. The paper proposes a classification of flexibility types and provides an overview of design strategies that have been adopted so far to enable different types of flexibility. Finally, it offers a conceptual framework that can support designers to evaluate specific types of flexibility in early-stage assessments of novel chemical processes.
Jisiwei Luo, Jonathan Moncada and Andrea Ramirez
-
The application of a polymer electrolyte membrane (PEM) electrolytic cell for continuous conversion of nitrate, one of the contaminants in water, to ammonia at the cathode was explored in the present work. Among carbon-supported metal (Cu, Ru, Rh and Pd) electrocatalysts, the Ru-based catalyst showed the best performance. By suppressing the competing hydrogen evolution reaction at the cathode, it was possible to reach 94 % faradaic efficiency for nitrate reduction towards ammonium. It was important to match the rate of the anodic reaction with the cathodic reaction to achieve high faradaic efficiency. By recirculating the effluent stream, 93 % nitrate conversion was achieved in 8 h of constant current electrolysis at 10 mA cm−2 current density. The presented approach offers a promising path towards precious NH3 production from nitrate-containing water that needs purification or can be obtained after capture of gaseous NOx pollutants into water, leading to waste-to-value conversion.
Sorin Bunea, Kevin Clemens, Prof.dr. Atsushi Urakawa
-
Low temperature electrolysis brings the possibility of achieving the production of fuels and chemical feedstocks without any carbon footprint. Power electronic converters are vital components of future electrolyser systems in terms of overall cost and efficiency. This paper presents the current state of art in power electronics for low temperature electrolysis and all the major steps of designing an electrolysis system are discussed from the modeling of electrolysers to the system architecture. The most promising routes are pointed out and backed up with results from both experimental and theoretical studies found in the literature.
Cristian Pascalau, Thiago B. Soeiro, Nils H. van der Blij, Pavol Bauer
-
Direct electrochemical reduction of CO2 to C2 products such as ethylene is more efficient in alkaline media, but it suffers from parasitic loss of reactants due to (bi)carbonate formation. A two-step process where the CO2 is first electrochemically reduced to CO and subsequently converted to desired C2 products has the potential to overcome the limitations posed by direct CO2 electroreduction. In this study, we investigated the technical and economic feasibility of the direct and indirect CO2 conversion routes to C2 products. For the indirect route, CO2 to CO conversion in a high temperature solid oxide electrolysis cell (SOEC) or a low temperature electrolyzer has been considered. The product distribution, conversion, selectivities, current densities, and cell potentials are different for both CO2 conversion routes, which affects the downstream processing and the economics. A detailed process design and techno-economic analysis of both CO2 conversion pathways are presented, which includes CO2 capture, CO2 (and CO) conversion, CO2 (and CO) recycling, and product separation. Our economic analysis shows that both conversion routes are not profitable under the base case scenario, but the economics can be improved significantly by reducing the cell voltage, the capital cost of the electrolyzers, and the electricity price. For both routes, a cell voltage of 2.5 V, a capital cost of $10,000/m2, and an electricity price of <$20/MWh will yield a positive net present value and payback times of less than 15 years. Overall, the high temperature (SOEC-based) two-step conversion process has a greater potential for scale-up than the direct electrochemical conversion route. Strategies for integrating the electrochemical CO2/CO conversion process into the existing gas and oil infrastructure are outlined. Current barriers for industrialization of CO2 electrolyzers and possible solutions are discussed as well.
Mahinder Ramdin, Bert De Mot, Andrew R. T. Morrison, Tom Breugelmans, Leo J. P. van den Broeke, J. P. Martin Trusler, Ruud Kortlever, Wiebren de Jong, Othonas A. Moultos, Penny Xiao, Paul A. Webley, and Thijs J. H. Vlugt
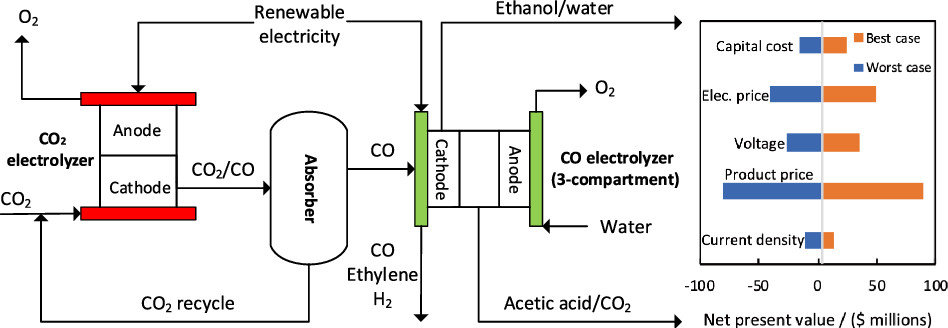
-
Electrochemical CO2 conversion into fuels or chemicals and CO2 capture from point or dilute sources are two important processes to address the gigaton challenges in reducing greenhouse gas emissions. Both CO2 capture and electrochemical CO2 conversion are energy intensive, and synergistic coupling between the two processes can improve the energy efficiency of the system and reduce the cost of the reduced products, via eliminating the CO2 transport and storage or eliminating the capture media regeneration and molecular CO2 release. We consider three different levels to couple electrochemical CO2 reduction with CO2 capture: independent (Type-I), subsequent (Type-II) and fully integrated (Type-III) capture and conversion processes. We focus on Type-II and Type-III configurations and illustrate potential coupling routes of different capture media, which include amine-based solutions and direct carbamate reduction, redox active carriers, aqueous carbonate and bicarbonate solutions, ionic liquids CO2 capture and conversion mediated by covalent organic frameworks.
Ian Sullivan, Andrey Goryachev, Ibadillah A. Digdaya, Xueqian Li, Harry A. Atwater, David A. Vermaas & Chengxiang Xiang
-
Direct electrochemical reduction of CO2 to C2 products such as ethylene is more efficient in alkaline media, but it suffers from parasitic loss of reactants due to (bi)carbonate formation. A two-step process where the CO2 is first electrochemically reduced to CO and subsequently converted to desired C2 products has the potential to overcome the limitations posed by direct CO2 electroreduction. In this study, we investigated the technical and economic feasibility of the direct and indirect CO2 conversion routes to C2 products. For the indirect route, CO2 to CO conversion in a high temperature solid oxide electrolysis cell (SOEC) or a low temperature electrolyzer has been considered. The product distribution, conversion, selectivities, current densities, and cell potentials are different for both CO2 conversion routes, which affects the downstream processing and the economics. A detailed process design and techno-economic analysis of both CO2 conversion pathways are presented, which includes CO2 capture, CO2 (and CO) conversion, CO2 (and CO) recycling, and product separation. Our economic analysis shows that both conversion routes are not profitable under the base case scenario, but the economics can be improved significantly by reducing the cell voltage, the capital cost of the electrolyzers, and the electricity price. For both routes, a cell voltage of 2.5 V, a capital cost of $10,000/m2, and an electricity price of <$20/MWh will yield a positive net present value and payback times of less than 15 years. Overall, the high temperature (SOEC-based) two-step conversion process has a greater potential for scale-up than the direct electrochemical conversion route. Strategies for integrating the electrochemical CO2/CO conversion process into the existing gas and oil infrastructure are outlined. Current barriers for industrialization of CO2 electrolyzers and possible solutions are discussed as well.
Mahinder Ramdin, Bert De Mot, Andrew R. T. Morrison, Tom Breugelmans, Leo J. P. van den Broeke, J. P. Martin Trusler, Ruud Kortlever, Wiebren de Jong, Othonas A. Moultos, Penny Xiao, Paul A. Webley and Thijs J. H. Vlugt

-
Methanation is a potential large-scale option for CO2 utilization, and it is one of the solutions for decreasing carbon emission and production of synthetic green fuels. However, the CO2 conversion is limited by thermodynamics in conventional reaction conditions. However, around 100 % conversion can be obtained using sorption enhanced CO2 methanation according to Le Chatelier’s principle, where water is removed during the reaction using zeolite as a sorbent. In this work 5%Ni5A, 5%Ni13X, 5%NiL and 5%Ni2.5%Ce13X bifunctional materials with both catalytic and water adsorption properties were tested in a fixed bed reactor. The overall performance of the bifunctional materials decreased on going from 5%Ni2.5%Ce13X, 5%Ni13X, 5%Ni5A, to 5%NiL. The CO2 conversion and CH4 selectivity were approaching 100 % during prolonged stability testing in a 100 reactive adsorption – desorption cycles test for 5%Ni2.5%Ce13X, and only a slight decrease of the water uptake capacity was observed.
Liangyuan Wei, Hamza Azad, Wim Haije, Henrik Grenman and Wiebren de Jong
-
Nowadays, Sn-based electrocatalysts for the electrochemical CO2 reduction reaction (eCO2RR) toward formic acid have been reported to reach industrially relevant current densities and Faradaic efficiencies approaching 100%. However, electrocatalyst stability remains inadequate and appears to be a crucial piece to the puzzle, as lifetimes in the range of several thousands of hours should be reached for practical application and economic viability. Here, we provide insights into stability issues related to Sn-based electrocatalysts and electrolyzers for formic acid production. By determining the chemical and physical phenomena that occur during the electrochemical reduction reaction on the surface and bulk of Sn-based catalysts, we intend to elucidate the most common degradation mechanisms that impair long-term electrocatalytic activity of these catalysts. Moreover, highlighting the importance of correctly selected process conditions and an optimized reactor design allows us to unveil all necessary aspects for a stable Sn-based eCO2RR toward formic acid.
Kevin Van Daele, Bert De Mot, Marilia Pupo, Nick Daems, Deepak Pant, Ruud Kortlever and Tom Breugelmans

-
Electrochemical CO2 capture technologies are gaining attention due to their flexibility, their ability to address decentralized emissions (e.g., ocean and atmosphere) and their fit in an electrified industry. In the present work, recent progress made in electrochemical CO2 capture is reviewed. The majority of these methods rely on the concept of “pH-swing” and the effect it has on the CO2 hydration/dehydration equilibrium. Through a pH-swing, CO2 can be captured and recovered by shifting the pH of a working fluid between acidic and basic pH. Such swing can be applied electrochemically through electrolysis, bipolar membrane electrodialysis, reversible redox reactions and capacitive deionization. In this review, we summarize main parameters governing these electrochemical pH-swing processes and put the concept in the framework of available worldwide capture technologies. We analyse the energy efficiency and consumption of such systems, and provide recommendations for further improvements. Although electrochemical CO2 capture technologies are rather costly compared to the amine based capture, they can be particularly interesting if more affordable renewable electricity and materials (e.g., electrode and membranes) become widely available. Furthermore, electrochemical methods have the ability to (directly) convert the captured CO2 to value added chemicals and fuels, and hence prepare for a fully electrified circular carbon economy.
R. Sharifian, R. M. Wagterveld, I. A. Digdaya, C. Xiang and D. A. Vermaas
-
Advancing reaction rates for electrochemical CO2 reduction in membrane electrode assemblies (MEAs) have boosted the promise of the technology while exposing new shortcomings. Among these is the maximum utilization of CO2, which is capped at 50% (CO as targeted product) due to unwanted homogeneous reactions. Using bipolar membranes in an MEA (BPMEA) has the capability of preventing parasitic CO2 losses, but their promise is dampened by poor CO2 activity and selectivity. In this work, we enable a 3-fold increase in the CO2 reduction selectivity of a BPMEA system by promoting alkali cation (K+) concentrations on the catalyst’s surface, achieving a CO Faradaic efficiency of 68%. When compared to an anion exchange membrane, the cation-infused bipolar membrane (BPM) system shows a 5-fold reduction in CO2 loss at similar current densities, while breaking the 50% CO2 utilization mark. The work provides a combined cation and BPM strategy for overcoming CO2 utilization issues in CO2 electrolyzers.
Kailun Yang, Mengran Li, Siddhartha Subramanian, Marijn A. Blommaert, Wilson A. Smith and Thomas Burdyny
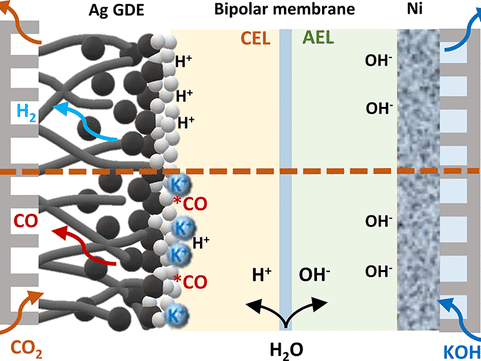
-
The production of value added C1 and C2 compounds within CO2 electrolyzers has reached sufficient catalytic performance that system and process performance – such as CO2 utilization – have come more into consideration. Efforts to assess the limitations of CO2 conversion and crossover within electrochemical systems have been performed, providing valuable information to position CO2 electrolyzers within a larger process. Currently missing, however, is a clear elucidation of the inevitable trade-offs that exist between CO2 utilization and electrolyzer performance, specifically how the faradaic efficiency of a system varies with CO2 availability. Such information is needed to properly assess the viability of the technology. In this work, we provide a combined experimental and 3D modelling assessment of the trade-offs between CO2 utilization and selectivity at 200 mA cm−2 within a membrane-electrode assembly CO2 electrolyzer. Using varying inlet flow rates we demonstrate that the variation in spatial concentration of CO2 leads to spatial variations in faradaic efficiency that cannot be captured using common ‘black box’ measurement procedures. Specifically, losses of faradaic efficiency are observed to occur even at incomplete CO2 consumption (80%). Modelling of the gas channel and diffusion layers indicated that at least a portion of the H2 generated is considered as avoidable by proper flow field design and modification. The combined work allows for a spatially resolved interpretation of product selectivity occurring inside the reactor, providing the foundation for design rules in balancing CO2 utilization and device performance in both lab and scaled applications.
Siddhartha Subramanian, Joost Middelkoop and Thomas Burdyny
Read the article
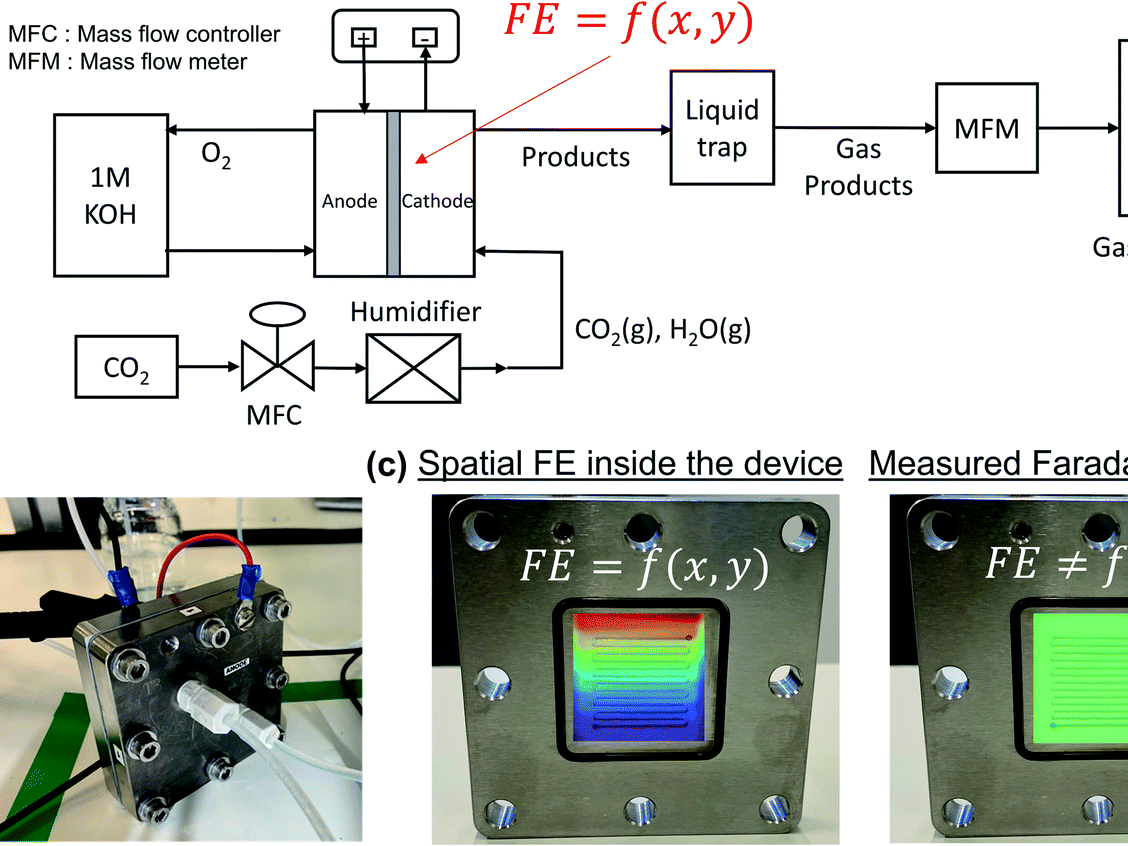
-
The electrochemical reduction of CO2 on planar electrodes is limited by its prohibitively low diffusivity and solubility in water. Gas-diffusion electrodes (GDEs) can be used to reduce these limitations, and facilitate current densities orders of magnitude higher than the limiting current densities of planar electrodes. These improvements are accompanied by increased variation in the local environment within the cathode, with significant effect on Faradaic efficiency. By developing a simple and freely available analytical model of a cathodic catalyst layer configured for the production of CO, we investigate the relationships between electrode reaction kinetics, cell operation conditions, catholyte composition and cell performance. Analytical methods allow us to cover parameter ranges that are intractable for numerical and experimental studies. We validate our findings against experimental and numerical results and provide a derivation and implementation of the analytical model.
J.W.Blake, J.T.Padding and J.W.Haverkort
-
Direct electrochemical nitrogen reduction holds the promise of enabling the production of carbon emission-free ammonia, which is an important intermediate in the fertilizer industry and a potential green energy carrier. Here we show a strategy for ambient condition ammonia synthesis using a hydrogen permeable nickel membrane/electrode that spatially separates the electrolyte and hydrogen reduction side from the dinitrogen activation and hydrogenation sites. Gaseous ammonia is produced catalytically in the absence of electrolyte via hydrogenation of adsorbed nitrogen by electrochemically permeating atomic hydrogen from water reduction. Dinitrogen activation at the polycrystalline nickel surface is confirmed with 15N2 isotope labeling experiments, and it is attributed to a Mars–van Krevelen mechanism enabled by the formation of N-vacancies upon hydrogenation of surface nitrides. We further show that gaseous hydrogen does not hydrogenate the adsorbed nitrogen, strengthening the benefit of having an atomic hydrogen permeable electrode. The proposed approach opens new directions toward green ammonia.
Davide Ripepi, Riccardo Zaffaroni, Herman Schreuders, Bart Boshuizen and Fokko M. Mulder

-
The electrochemical reduction of carbon dioxide (CO2RR) requires access to ample gaseous CO2 and liquid water to fuel reactions at high current densities for industrial-scale applications. Substantial improvement of the CO2RR rate has largely arisen from positioning the catalyst close to gas–liquid interfaces, such as in gas-diffusion electrodes. These requirements add complexity to an electrode design that no longer consists of only a catalyst but also a microporous and nanoporous network of gas–liquid–solid interfaces of the electrode. In this three-dimensional structure, electrode wettability plays a pivotal role in the CO2RR because the affinity of the electrode surface by water impacts the observed electrode reactivity, product selectivity, and long-term stability. All these performance metrics are critical in an industrial electrochemical process. This review provides an in-depth analysis of electrode wettability's role in achieving an efficient, selective, and stable CO2RR performance. We first discuss the underlying mechanisms of electrode wetting phenomena and the foreseen ideal wetting conditions for the CO2RR. Then we summarize recent advances in improving cathode performance by altering the wettability of the catalyst layer of gas-diffusion electrodes. We conclude the review by discussing the current challenges and opportunities to develop efficient and selective cathodes for CO2RR at industrially relevant rates. The insights generated from this review could also benefit the advancement of other critical electrochemical processes that involve multiple complex flows in porous electrodes, such as electrochemical reduction of carbon monoxide, oxygen, and nitrogen.
Mengran Li, Mohamed Nazmi Idros, Yuming Wu, Thomas Burdyny, Sahil Garg, Xiu Song Zhao, Geoff Wang and Thomas E. Rufford
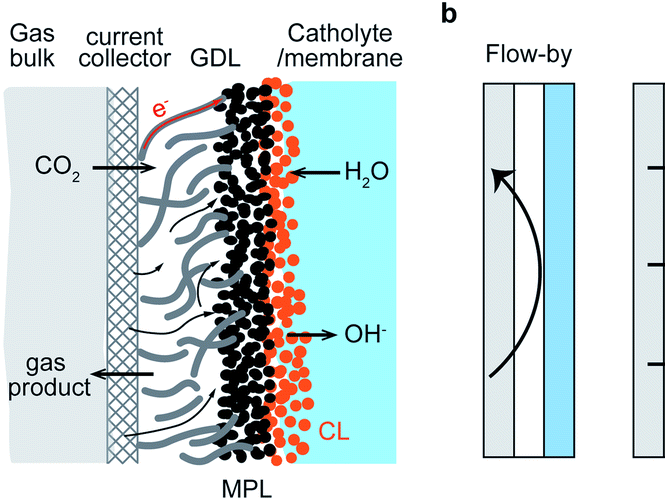
-
Bipolar membranes (BPMs) are gaining interest in energy conversion technologies. These membranes are composed of cation- and anion-exchange layers, with an interfacial layer in between. This gives the freedom to operate in different conditions (pH, concentration, composition) at both sides. Such membranes are used in two operational modes, forward and reverse bias. BPMs have been implemented in various electrochemical applications, like water and CO2 electrolyzers, fuel cells, and flow batteries, while BPMs are historically designed for acid/base production. Therefore, current commercial BPMs are not optimized, as the conditions change per application. Although the ideal BPM has highly conductive layers, high water dissociation kinetics, long lifetime, and low ion crossover, each application has its own priorities to be competitive in its field. We describe the challenges and requirements for future BPMs, and identify existing developments that can be leveraged to develop BPMs toward the scale of practical applications.
Marijn A. Blommaert, David Aili, Ramato Ashu Tufa, Qingfeng Li, Wilson A. Smith and David A. Vermaas
-
Oil palm empty fruit bunch (OPEFB) is an abundant waste that is commonly incinerated, causing environmental pollution. In this study, an alternative waste management approach was investigated to produce value-added syngas from OPEFB using solar steam gasification. The three operating variables were temperature (1100–1300 °C), H2O/OPEFB molar ratio (1.7–2.9), and OPEFB flowrate (0.8–1.8 g/min). Central composite design (CCD) was conducted to investigate and optimise the effects of these operating variables on H2/CO molar ratio and solar to fuel energy conversion efficiency (ηsolar to fuel). The findings revealed that all investigated operating variables were significant. Experimentally, the highest H2/CO molar ratio (1.6) was obtained at 1300 °C, H2O/OPEFB molar ratio of 2.9, and OPEFB flowrate of 1.8 g/min, with a high carbon conversion reaching 95.1%. Results from CCD analysis showed that a higher H2/CO molar ratio (above 1.8) could be reached at 1200 °C, H2O/OPEFB molar ratio of ≥3.0, and OPEFB flowrate of ≥2.0 g/min. The maximum ηsolar to fuel of 19.6% was achieved at 1200 °C, H2O/OPEFB molar ratio of 1.3, and OPEFB flowrate of 1.3 g/min, whereby a favourable energy upgrade factor (1.2) was achieved. The statistical model showed adequacy to predict H2/CO molar ratio.
Saqr A.A. Al-Muraisy, Lais Americo Soares, Srirat Chuayboon, Shahrul Bin Ismail , Stéphane Abanades, Jules B. van Lier and Ralph E.F. Lindeboom
-
CO2 electroreduction provides a route to convert waste emissions into chemicals such as ethylene (C2H4). However, the direct transformation of CO2-to-C2H4 suffers from CO2 loss to carbonate, consuming up to 72% of energy input. A cascade approach—coupling a solid-oxide CO2-to-CO electrochemical cell (SOEC) with a CO-to-C2H4 membrane electrode assembly (MEA)—would eliminate CO2 loss to carbonate. However, this approach requires a CO-to-C2H4 MEA with energy efficiency well beyond demonstrations to date. Focusing on the MEA, we find that an N-tolyl substituted tetrahydro-bipyridine film improves the stabilization of key reaction intermediates, while an SSC ionomer enhances CO transport to the Cu surface, enabling a C2H4 faradaic efficiency of 65% at 150 mA cm−2 for 110 h. Demonstrating a cascade SOEC-MEA approach, we achieve CO2-to-C2H4 with a ~48% reduction in energy intensity compared with the direct route. We further reduce the energy intensity by coupling CO electroreduction (CORR) with glucose electrooxidation.
A Ozden, Y Wang, Li F, Luo M, Sisler J, Thevenon A, Rosas-Hernández A, Burdyny T, Lum Y, Yadegari H, Agapie T, Peters JC, Sargent EH, Sinton D.
-
Sub-nanometer zeolite 13X-supported Ni-ceria catalysts were synthesized for CO2 methanation. XRD and SEM results show the structure and morphology of 13X zeolite after impregnation and calcination. Ce loading affected the catalysts’ metal dispersion, reducibility, basicity and acidity, and thence their activity and selectivity. STEM-EDX elemental mappings showed that Ce and Ni are predominantly highly dispersed. Ce has a positive effect on the reduction of NiO and leads to a relatively high number of medium basic sites with a low Ce loading. Highly stable 5%Ni2.5%Ce13X had high activity and nearly 100% CH4 selectivity in CO2 methanation at 360 °C, which is mainly due to the high dispersion of metals and relatively high amount of medium basic sites. It can be inferred that this catalyst synthesis strategy has great potential for good catalyst dispersion, since metal uptake by the zeolite is selective for the metal citrate complexes in solution.
Liangyuan Wei, Henrik Grénman, Wim Haije, Narendra Kumar, Atte Aho, Kari Eränen, Liangfu Wei, Wiebren de Jong
-
Zeolite 13X and 5A supported Ni catalysts were synthesized for CO2 methanation using the evaporation impregnation method. The influence of using different Ni precursors (nitrate, citrate, and acetate) as well as calcination temperatures on the catalyst properties and performance were investigated. XRD, SEM-EDX, TEM, STEM-EDX, N2 physisorption, H2-TPR, TPD-NH3 and TG/DTA were used for detailed characterization of the catalysts. The parent structure of the zeolites did not change during catalyst synthesis. Using nickel citrate and acetate resulted in smaller NiO particle size compared to nitrate. STEM-EDX results showed that all the Ni-precursor complexes entered more efficiently the 13X zeolite structure, which is mainly due to steric hindrance resulting from the smaller pore size of 5A. Methanation experiments revealed that the 13X catalysts synthesized using nickel citrate (5% Ni) displayed clearly higher activity, compared to the catalysts synthesized using nickel nitrate or nickel acetate. A 79% conversion at 320 °C was obtained with 100% selectivity towards CH4 and the catalyst showed excellent stability during 200 h testing. Overall, it can be concluded that the Ni precursor significantly influences the physico-chemical characteristics and catalytic properties of Ni 13X and Ni 5A zeolite catalysts in CO2 methanation: complex size and pore size matter.
Liangyuan Wei, Wim Haije, Narendra Kumar, Janne Peltonen, Markus Peurla, Henrik Grenman, Wiebren de Jong
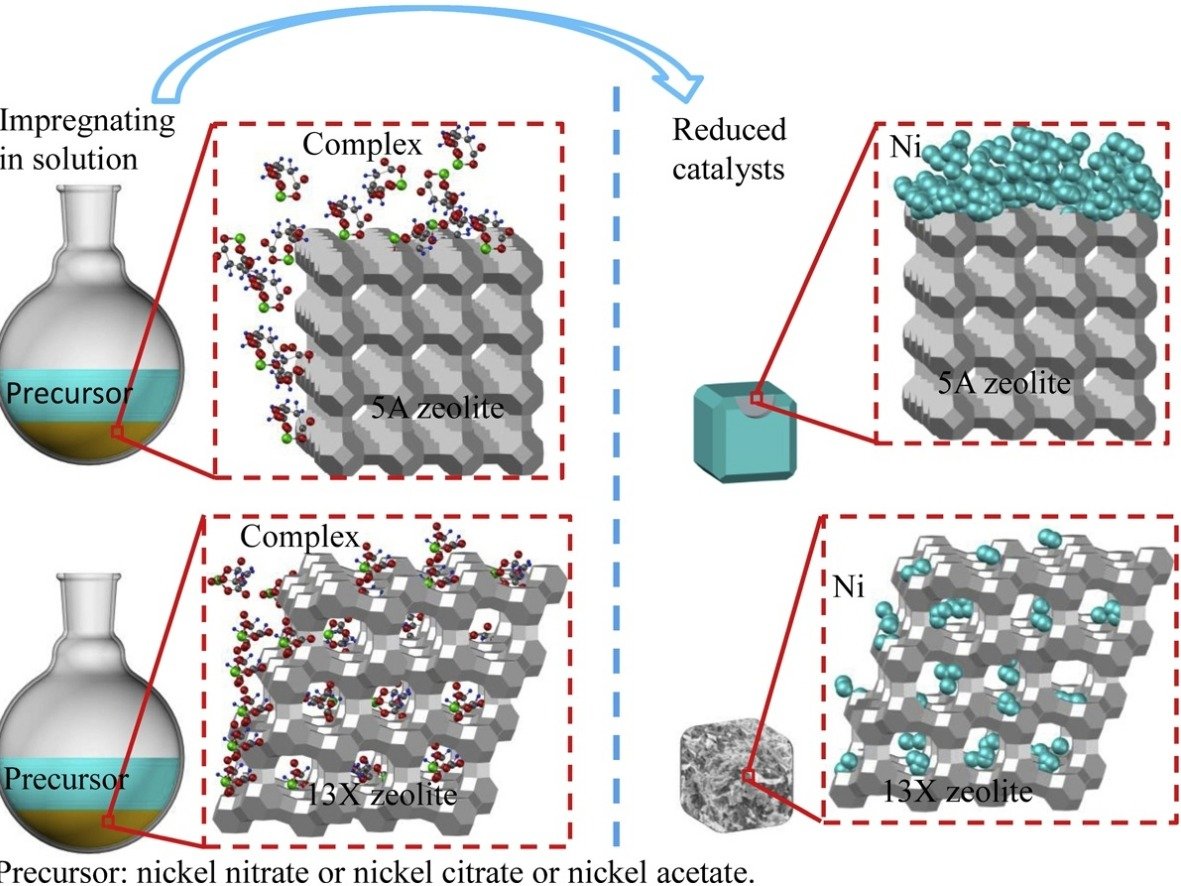
-
Methanation is a potential large-scale option for CO2 utilization, and it is one of the solutions for decreasing carbon emission and production of synthetic green fuels. However, the CO2 conversion is limited by thermodynamics in conventional reaction conditions. However, around 100 % conversion can be obtained using sorption enhanced CO2 methanation according to Le Chatelier’s principle, where water is removed during the reaction using zeolite as a sorbent. In this work 5%Ni5A, 5%Ni13X, 5%NiL and 5%Ni2.5%Ce13X bifunctional materials with both catalytic and water adsorption properties were tested in a fixed bed reactor. The overall performance of the bifunctional materials decreased on going from 5%Ni2.5%Ce13X, 5%Ni13X, 5%Ni5A, to 5%NiL. The CO2 conversion and CH4 selectivity were approaching 100 % during prolonged stability testing in a 100 reactive adsorption – desorption cycles test for 5%Ni2.5%Ce13X, and only a slight decrease of the water uptake capacity was observed.
Drs. Liangyuan Wei, Drs. Hamza Azad, Dr. Wim Haije, Prof.dr.ir. Wiebren de Jong
-
Due to over- and under-supply, renewable energy sources can have a major impact on the electricity market and cause fluctuating electricity prices. In their laboratory, dr. Audrey Iranzo and prof.dr. Fokko Mulder developed a Ni-Fe layered double hydroxide for use as a positive electrode in the integrated nickel-iron battery and alkaline electrolyser.
Dr. Audrey Iranzo and prof.dr. Fokko Mulder
-
The deployment of gas diffusion electrodes (GDEs) for the electrochemical CO2 reduction reaction (CO2RR) has enabled current densities an order of magnitude greater than those of aqueous H cells. The gains in production, however, have come with stability challenges due to rapid flooding of GDEs, which frustrate both laboratory experiments and scale-up prospects. Here, we investigate the role of carbon gas diffusion layers (GDLs) in the advent of flooding during CO2RR, finding that applied potential plays a central role in the observed instabilities. Electrochemical characterization of carbon GDLs with and without catalysts suggests that the high overpotential required during electrochemical CO2RR initiates hydrogen evolution on the carbon GDL support. These potentials impact the wetting characteristics of the hydrophobic GDL, resulting in flooding that is independent of CO2RR. Findings from this work can be extended to any electrochemical reduction reaction using carbon-based GDEs (CORR or N2RR) with cathodic overpotentials of less than −0.65 V versus a reversible hydrogen electrode.
Yang K., Kas R., Smith WA., Burdyny T.
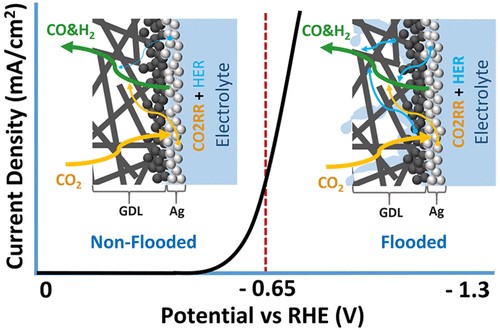
-
Electrolysis-assisted nitrate (NO3−) reduction is a promising approach for its conversion to harmless N2 from waste, ground, and drinking water due to the possible process simplicity by in-situ generation of H2/H/H+ by water electrolysis and to the flexibility given by tunable redox potential of electrodes. This work explores the use of a polymer electrolyte membrane (PEM) electrochemical cell for electrolysis-assisted nitrate reduction using SnO2-supported metals as the active cathode catalysts. Effects of operation modes and catalyst materials on nitrate conversion and product selectivity were studied. The major challenge of product selectivity, namely complete suppression of nitrite (NO2−) and ammonium (NH4+) ion formation, was tackled by combining with simultaneous photocatalytic oxidation to drive the overall reaction towards N2 formation.
Dr. Jordi Ampurdanés, Sorin Bunea, Prof.dr. Atsushi Urakawa
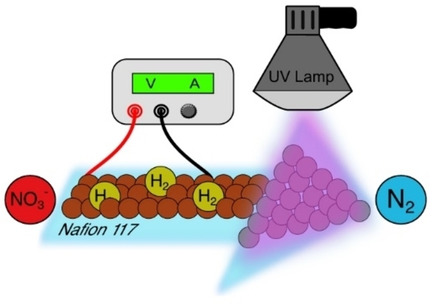
-
Electrochemical CO2 electrolysis to produce hydrocarbon fuels or material feedstocks offers a renewable alternative to fossilized carbon sources. Gas-diffusion electrodes (GDEs), composed of solid electrocatalysts on porous supports positioned near the interface of a conducting electrolyte and CO2 gas, have been able to demonstrate the substantial current densities needed for future commercialization. These higher reaction rates have often been ascribed to the presence of a three-phase interface, where solid, liquid, and gas provide electrons, water, and CO2, respectively. Conversely, mechanistic work on electrochemical reactions implicates a fully two-phase reaction interface, where gas molecules reach the electrocatalyst’s surface by dissolution and diffusion through the electrolyte. Because the discrepancy between an atomistic three-phase versus two-phase reaction has substantial implications for the design of catalysts, gas-diffusion layers, and cell architectures, the nuances of nomenclatures and governing phenomena surrounding the three-phase-region require clarification. Here we outline the macro, micro, and atomistic phenomena occurring within a gas-diffusion electrode to provide a focused discussion on the architecture of the often-discussed three-phase region for CO2 electrolysis. From this information, we comment on the outlook for the broader CO2 electroreduction GDE cell architecture.
Nesbitt NT., Burdyny T., Simonson H., Salvatore D., Bohra D., Kas R., Smith WA.
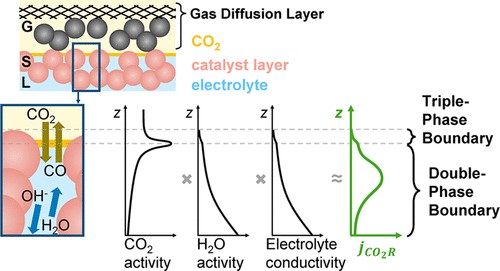
-
X-ray absorption spectroscopy (XAS) offers the unique possibility to study metal electrocatalysts such as silver and copper while they are performing electrochemical carbon dioxide (CO2) reduction. In this work, we present an approach to perform operando XAS experiments on an electrochemical cell performing CO2 reduction with a gas diffusion electrode (GDE) as cathode. The experimental set-up, advantages and drawbacks, XAS data analysis, and XAS theory are discussed. Results on copper and silver GDEs obtained through the presented procedures are then presented and discussed. Structural and compositional catalyst data acquired under operando conditions can help further density functional theory calculations, and catalytic, and systems studies on CO2 reduction. Structural and compositional data including crystallite size were obtained while performing high current density (up to 200 mA cm−2) CO2 reduction. On the silver catalysts at higher than 100 mA cm−2 applied current density, a Ag–X contribution was found and is ascribed to Ag–O. For both silver and copper, the XAS experiments revealed that the crystallite size of the ex situ samples is smaller than the samples during CO2 reduction. Furthermore, metal particle size polydispersity was found in the silver catalysts by comparing the obtained coordination numbers with theoretical values. The operando EXAFS data was of such high quality (k: 3–14 Å−1) that four shells could be fitted. The value of combining ex situ material characterisation and electrocatalyst performance data with operando XAS experiments is discussed and found to be of great importance to further CO2 reduction research.
Firet NJ., Burdyny T., Nesbitt NT., Chandrashekar S., Longo A., Smith WA.
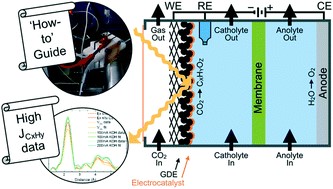
-
Despite substantial progress in the electrochemical conversion of CO2 into value-added chemicals, the translation of fundamental studies into commercially relevant conditions requires additional efforts. Here, we study the catalytic properties of tailored Cu nanocatalysts under commercially relevant current densities in a gas-fed flow cell. We demonstrate that their facet-dependent selectivity is retained in this device configuration with the advantage of further suppressing hydrogen production and increasing the faradaic efficiencies toward the CO2 reduction products compared to a conventional H-cell. The combined catalyst and system effects result in state-of-the art product selectivity at high current densities (in the range 100–300 mA/cm2) and at relatively low applied potential (as low as −0.65 V vs RHE). Cu cubes reach an ethylene selectivity of up to 57% with a corresponding mass activity of 700 mA/mg, and Cu octahedra reach a methane selectivity of up to 51% with a corresponding mass activity of 1.45 A/mg in 1 M KOH.
De Gregorio GL., Burdyny T., Loiudice A., Iyengar P., Smith WA., Buonsanti R.

-
Electrochemical CO2 reduction has received an increased amount of interest in the last decade as a promising avenue for storing renewable electricity in chemical bonds. Despite considerable progress on catalyst performance using nanostructured electrodes, the sensitivity of the reaction to process conditions has led to debate on the origin of the activity and high selectivity. Additionally, this raises questions on the transferability of the performance and knowledge to other electrochemical systems. At its core, the discrepancy is primarily a result of the highly porous nature of nanostructured electrodes, which are vulnerable to both mass transport effects and structural changes during the electrolysis. Both effects are not straightforward to identify and difficult to decouple. Despite the susceptibility of nanostructured electrodes to mass transfer limitations, we highlight that nanostructured silver electrodes exhibit considerably higher activity when normalized to the electrochemically active surface in contrast to gold and copper electrodes. Alongside, we provide a discussion on how active surface area and thickness of the catalytic layer itself can influence the onset potential, selectivity, stability, activity and mass transfer inside and outside of the three dimensional catalyst layer. Key parameters and potential solutions are highlighted to decouple mass transfer effects from the measured activity in electrochemical cells utilizing CO2 saturated aqueous solutions.
Kas K., Yang K., Bohra D., Kortlever R., Burdyny T., Smith WA.

-
The environment of a CO2 electroreduction (CO2ER) catalyst is intimately coupled with the surface reaction energetics and is therefore a critical aspect of the overall system performance. The immediate reaction environment of the electrocatalyst constitutes the electrical double layer (EDL) which extends a few nanometers into the electrolyte and screens the surface charge density. In this study, we resolve the species concentrations and potential profiles in the EDL of a CO2ER system by self-consistently solving the migration, diffusion and reaction phenomena using the generalized modified Poisson–Nernst–Planck (GMPNP) equations which include the effect of volume exclusion due to the solvated size of solution species. We demonstrate that the concentration of solvated cations builds at the outer Helmholtz plane (OHP) with increasing applied potential until the steric limit is reached. The formation of the EDL is expected to have important consequences for the transport of the CO2 molecule to the catalyst surface. The electric field in the EDL diminishes the pH in the first 5 nm from the OHP, with an accumulation of protons and a concomitant depletion of hydroxide ions. This is a considerable departure from the results obtained using reaction-diffusion models where migration is ignored. Finally, we use the GMPNP model to compare the nature of the EDL for different alkali metal cations to show the effect of solvated size and polarization of water on the resultant electric field. Our results establish the significance of the EDL and electrostatic forces in defining the local reaction environment of CO2 electrocatalysts.
Bohra D., Chaudhry JH., Burdyny T., Pidko EA., Smith WA.
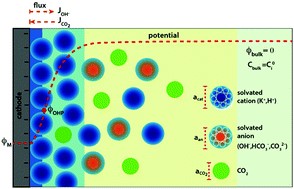
-
A successful transition of our global energy system will require changes to our society and industry. One of these changes is the production of electricity from renewable energy sources, which can be directly used today and in our increasingly electrified future infrastructure. The generated green electricity is also foreseen to be essential in “Power-to-X” technologies, where ambient molecules such as H2O, CO2, and N2 are converted into dense-energy carriers.
Here, we provide an example of the combined technologies needed to make Power-to-X into an industry capable of influencing the global energy system (~TW). Using CO2 electrolyzers as a central technology in the production of green methanol (MeOH), we discuss the integration of direct CO2 air capture with CO2 and H2O electrolyzers and a traditional MeOH synthesis step. The resulting analysis provides tangible scales for CO2 electrolyzers at an early research stage and perspectives on a future industry powered by dilute solar energy resources.
Smith WA., Burdyny T., Vermaas D., Geerlings H.
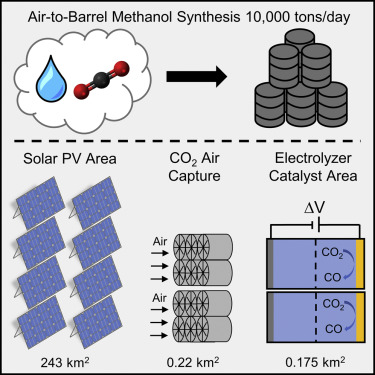
-
Electrocatalytic CO2 reduction has the dual-promise of neutralizing carbon emissions in the near future, while providing a long-term pathway to create energy-dense chemicals and fuels from atmospheric CO2. The field has advanced immensely in recent years, taking significant strides towards commercial realization. Catalyst innovations have played a pivotal role in these advances, with a steady stream of new catalysts providing gains in CO2 conversion efficiencies and selectivities of both C1 and C2 products. Comparatively few of these catalysts have been tested at commercially-relevant current densities (∼200 mA cm−2) due to transport limitations in traditional testing configurations and a research focus on fundamental catalyst kinetics, which are measured at substantially lower current densities. A catalyst's selectivity and activity, however, have been shown to be highly sensitive to the local reaction environment, which changes drastically as a function of reaction rate. As a consequence of this, the surface properties of many CO2 reduction catalysts risk being optimized for the wrong operating conditions. The goal of this perspective is to communicate the substantial impact of reaction rate on catalytic behaviour and the operation of gas-diffusion layers for the CO2 reduction reaction. In brief, this work motivates high current density catalyst testing as a necessary step to properly evaluate materials for electrochemical CO2 reduction, and to accelerate the technology toward its envisioned application of neutralizing CO2 emissions on a global scale.
Burdyny T., Smith WA.
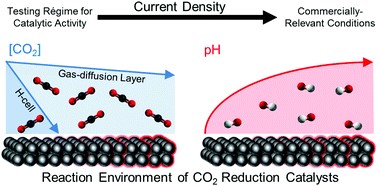
-
Electrocatalysis of carbon dioxide can provide a valuable pathway towards the sustainable production of chemicals and fuels from renewable electricity sources. One of the main challenges to enable this technology is to find suitable electrodes that can act as efficient, stable and selective CO2 reduction catalysts. Modified silver catalysts and in particular, catalysts electrochemically derived from silver-oxides, have shown great promise in this regard. Here, we use operando EXAFS analysis to study the differences in surface composition between a pure silver film and oxide-derived silver catalysts – a nanostructured catalyst with improved CO2 reduction performance. The EXAFS analysis reveals the presence of trace amounts of oxygen in the oxide-derived silver samples, with the measured oxygen content correlating well with experimental studies showing an increase in CO2 reduction reactivity towards carbon monoxide. The selectivity towards CO production also partially scales with the increased surface area, showing that the morphology, local composition and electronic structure all play important roles in the improved activity and selectivity of oxide-derived silver electrocatalysts. Earlier studies based on X-ray photoelectron spectroscopy (XPS) were not able to identify this oxygen, most likely because in ultra-high vacuum conditions, silver can self-reduce to Ag0, removing existing oxygen species. This operando EXAFS study shows the potential for in situ and operando techniques to probe catalyst surfaces during electrolysis and aid in the overall understanding of electrochemical systems.
Firet NJ., Blommaert MA., Burdyny T., Venugopal A., Bohra D., Longo A., Smith WA.